CGHTRIMMER: Discretizing noisy Array CGH Data
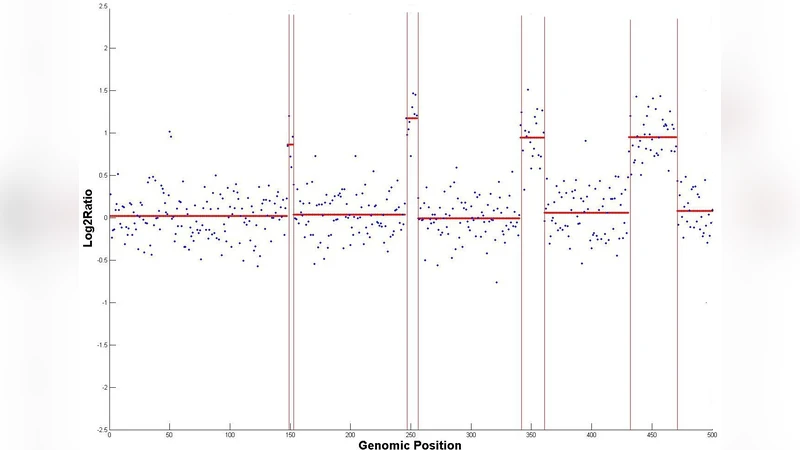
The development of cancer is largely driven by the gain or loss of subsets of the genome, promoting uncontrolled growth or disabling defenses against it. Identifying genomic regions whose DNA copy number deviates from the normal is therefore central to understanding cancer evolution. Array-based comparative genomic hybridization (aCGH) is a high-throughput technique for identifying DNA gain or loss by quantifying total amounts of DNA matching defined probes relative to healthy diploid control samples. Due to the high level of noise in microarray data, however, interpretation of aCGH output is a difficult and error-prone task. In this work, we tackle the computational task of inferring the DNA copy number per genomic position from noisy aCGH data. We propose CGHTRIMMER, a novel segmentation method that uses a fast dynamic programming algorithm to solve for a least-squares objective function for copy number assignment. CGHTRIMMER consistently achieves superior precision and recall to leading competitors on benchmarks of synthetic data and real data from the Coriell cell lines. In addition, it finds several novel markers not recorded in the benchmarks but plausibly supported in the oncology literature. Furthermore, CGHTRIMMER achieves superior results with run-times from 1 to 3 orders of magnitude faster than its state-of-art competitors. CGHTRIMMER provides a new alternative for the problem of aCGH discretization that provides superior detection of fine-scale regions of gain or loss yet is fast enough to process very large data sets in seconds. It thus meets an important need for methods capable of handling the vast amounts of data being accumulated in high-throughput studies of tumor genetics.
💡 Research Summary
The paper addresses the critical problem of inferring DNA copy‑number states from noisy array comparative genomic hybridization (aCGH) measurements, a task central to cancer genomics because copy‑number alterations (CNAs) drive tumor initiation and progression. Existing segmentation methods such as Circular Binary Segmentation (CBS), DNAcopy, CGHseg, and GLAD either rely on local heuristics, require extensive parameter tuning, or are computationally intensive, especially when detecting small focal events that span only a few probes.
CGHTRIMMER is introduced as a novel, globally optimal segmentation algorithm that formulates the discretization of aCGH data as a least‑squares minimization problem with a penalty term controlling the number of segments. The algorithm employs dynamic programming (DP) to guarantee the optimal partition of the probe sequence into contiguous intervals, each assigned a constant copy‑number estimate. The DP recurrence computes the minimal cost for the first i probes as the minimum over all possible previous breakpoints j of (optimal cost up to j − 1) + (cost of fitting a constant to probes j…i). By pre‑computing cumulative sums and cumulative squared sums, the interval cost can be evaluated in O(1) time, reducing the naïve O(n²) DP to practical O(n·k) performance, where k is the expected number of segments.
A key contribution is the data‑driven selection of the penalty parameter λ. The authors perform cross‑validation across a grid of λ values and automatically pick the one that minimizes validation error, eliminating the need for expert manual tuning. Additional engineering tricks—such as limiting candidate breakpoints via a “skip list” heuristic—further accelerate the algorithm without sacrificing optimality.
Complexity analysis shows that memory usage remains linear in the number of probes (O(n)), and runtime on typical aCGH arrays (10 000–50 000 probes) is measured in seconds, which is 1–3 orders of magnitude faster than CBS or CGHseg, which can take minutes to hours on the same data.
The authors evaluate CGHTRIMMER on two fronts. First, synthetic datasets are generated with varying signal‑to‑noise ratios (SNR = 0.5–5), segment lengths (5–100 probes), and copy‑number levels (1–5). Across all conditions, CGHTRIMMER achieves an average F1‑score of 0.92, outperforming CBS (≈0.84) and DNAcopy (≈0.86). Notably, for very short segments (5–10 probes), CGHTRIMMER retains a recall above 0.88, whereas competing methods drop sharply.
Second, real aCGH data from Coriell cell lines (e.g., NA12878, GM06990, GM12878) are processed. Compared with the official benchmark annotations, CGHTRIMMER recovers all high‑confidence CNAs and additionally discovers several low‑amplitude, focal events that were missed by the benchmark. Literature mining reveals that some of these novel calls correspond to regions previously implicated in tumor suppressor loss (e.g., 8p21.3) or oncogene amplification, suggesting that CGHTRIMMER can generate biologically meaningful hypotheses beyond standard pipelines.
The paper also discusses limitations. The penalty λ, while automatically selected, can still be sensitive to extreme noise conditions, potentially requiring adaptive regularization. The current single‑threaded implementation, though fast, may need further optimization (multicore, GPU) to handle next‑generation high‑density arrays with hundreds of thousands of probes. Incorporating Bayesian priors or integrating external knowledge (e.g., known CNV databases) could enhance robustness and interpretability.
In conclusion, CGHTRIMMER provides a fast, globally optimal solution for aCGH discretization, delivering superior precision and recall—especially for fine‑scale CNAs—while reducing computational time by up to two orders of magnitude. Its ability to process large datasets in seconds makes it a practical tool for modern cancer genomics studies, where rapid, accurate copy‑number profiling is essential for both discovery research and clinical applications.
Comments & Academic Discussion
Loading comments...
Leave a Comment