Flux Analysis in Process Models via Causality
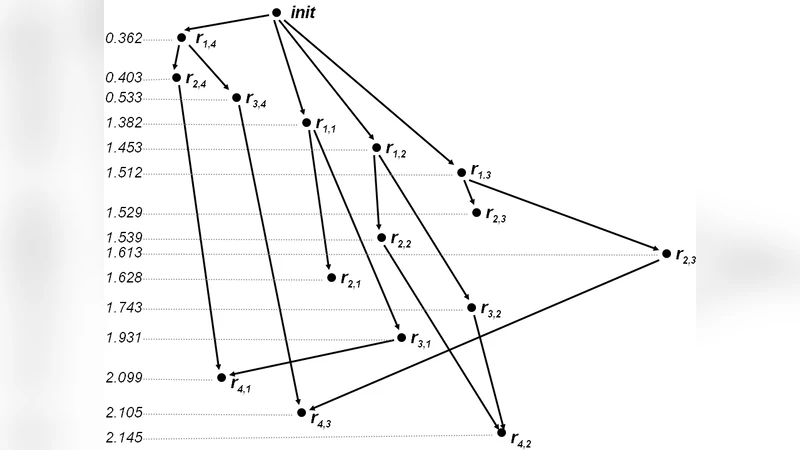
We present an approach for flux analysis in process algebra models of biological systems. We perceive flux as the flow of resources in stochastic simulations. We resort to an established correspondence between event structures, a broadly recognised model of concurrency, and state transitions of process models, seen as Petri nets. We show that we can this way extract the causal resource dependencies in simulations between individual state transitions as partial orders of events. We propose transformations on the partial orders that provide means for further analysis, and introduce a software tool, which implements these ideas. By means of an example of a published model of the Rho GTP-binding proteins, we argue that this approach can provide the substitute for flux analysis techniques on ordinary differential equation models within the stochastic setting of process algebras.
💡 Research Summary
The paper introduces a novel method for performing flux analysis on stochastic process‑algebra models of biochemical systems. Traditional flux analysis is usually carried out on deterministic ordinary‑differential‑equation (ODE) models, where the flow of metabolites is represented by averaged reaction rates. In contrast, stochastic process algebras (such as stochastic π‑calculus or Bio‑PEPA) generate individual simulation trajectories that exhibit variability in reaction ordering and concurrency, making ODE‑based techniques insufficient for capturing the full dynamics.
To bridge this gap, the authors exploit a well‑known correspondence between process‑algebraic specifications, Petri nets, and event structures—a formalism from concurrency theory that explicitly records causality and concurrency among events. The workflow proceeds as follows: a stochastic simulation produces a log of state transitions together with the reaction that triggered each transition. This log is parsed to construct a Petri net representation, where tokens model molecular species and transitions model reactions. Each transition is then mapped to an event in an event structure, and the causal dependencies between events (i.e., “event A must precede event B because B consumes a product of A”) are captured as a partial‑order graph. Unlike a simple time‑stamp sequence, this partial order reflects true logical precedence, preserving the concurrent nature of the underlying biochemical processes.
Two graph‑transformations are proposed to make the resulting structure amenable to analysis. The first is a transitive reduction that removes redundant edges, yielding a minimal causality graph. The second is an aggregation step that collapses multiple instances of the same reaction type (for example, all phosphorylations catalyzed by the same kinase) into a single meta‑node, thereby providing a higher‑level view of resource flow. Edge weights can be attached to indicate the number of occurrences or the probability of a particular causal link, enabling quantitative flux measurement alongside qualitative causality.
A software prototype implements this pipeline. It interfaces with existing stochastic simulators (e.g., SPiM, StochKit), extracts token movements, builds the event‑structure partial order using an open‑source library, applies the reduction and aggregation steps, and finally exports the graph in formats compatible with GraphViz and Cytoscape for interactive exploration.
The methodology is demonstrated on a published model of the Rho GTP‑binding protein cycle, a system that includes GTP/GDP exchange, GEF (guanine nucleotide exchange factor) activation, GAP (GTPase‑activating protein) mediated hydrolysis, and multiple feedback loops. When the authors apply their approach, they obtain a detailed causal map for each simulation run, revealing which reactions actually occurred together, which pathways were dominant, and how the introduction of an inhibitor reshapes the flow of GTP‑bound states. This level of detail is unattainable with ODE‑based flux analysis, which can only report average fluxes and cannot distinguish rare but biologically significant events.
The paper’s contributions are threefold. First, it establishes a rigorous theoretical link between stochastic process‑algebra semantics and event‑structure causality, thereby enabling flux analysis in a stochastic setting. Second, it provides concrete graph‑based transformations (transitive reduction and aggregation) that turn raw causal data into interpretable, hierarchical flux diagrams. Third, it validates the approach on a realistic biological case study and releases an open‑source tool that makes the technique accessible to the broader systems‑biology community.
Limitations are acknowledged. For very large models the number of events can explode, producing dense partial‑order graphs that challenge both visualization and computation. While the proposed reductions help, additional clustering or summarisation algorithms will be needed for scalability. Moreover, the current implementation processes single‑threaded simulation logs; extending it to handle parallel simulations or real‑time streaming data is an open research direction.
In summary, the authors present a compelling alternative to deterministic flux analysis by leveraging causality‑preserving event structures derived from stochastic process‑algebra simulations. Their method captures both quantitative flux magnitudes and the qualitative ordering of reactions, offering a richer, more faithful picture of biochemical dynamics. This work opens new avenues for model validation, hypothesis generation, and the integration of stochastic simulation data into the broader toolbox of systems biology.
Comments & Academic Discussion
Loading comments...
Leave a Comment