Monodisperse self-assembly in a model with protein-like interactions
We study the self-assembly behaviour of patchy particles with `protein-like’ interactions that can be considered as a minimal model for the assembly of viral capsids and other shell-like protein complexes. We thoroughly explore the thermodynamics and dynamics of self assembly as a function of the parameters of the model and find robust assembly of all target structures considered. Optimal assembly occurs in the region of parameter space where a free energy barrier regulates the rate of nucleation, thus preventing the premature exhaustion of the supply of monomers that can lead to the formation of incomplete shells. The interactions also need to be specific enough to prevent the assembly of malformed shells, but whilst maintaining kinetic accessibility. Free-energy landscapes computed for our model have a funnel-like topography guiding the system to form the target structure, and show that the torsional component of the interparticle interactions prevents the formation of disordered aggregates that would otherwise act as kinetic traps.
💡 Research Summary
The paper presents a comprehensive study of the self‑assembly of patchy particles endowed with “protein‑like” interactions, focusing on the formation of virus‑capsid‑like shells and other closed protein complexes. The authors extend the conventional patchy‑particle framework by explicitly incorporating a torsional component into the inter‑particle potential. In practice, each particle is modeled as a sphere decorated with a set of attractive patches; the interaction between two patches is governed by two tunable parameters: a binding strength ε that controls the depth of the attractive well, and a torsional stiffness κ that penalizes deviations from the preferred relative orientation of the patches. This addition captures the directional and rotational specificity that characterizes real protein‑protein interfaces, where the correct alignment of side‑chain contacts is essential for stable complex formation.
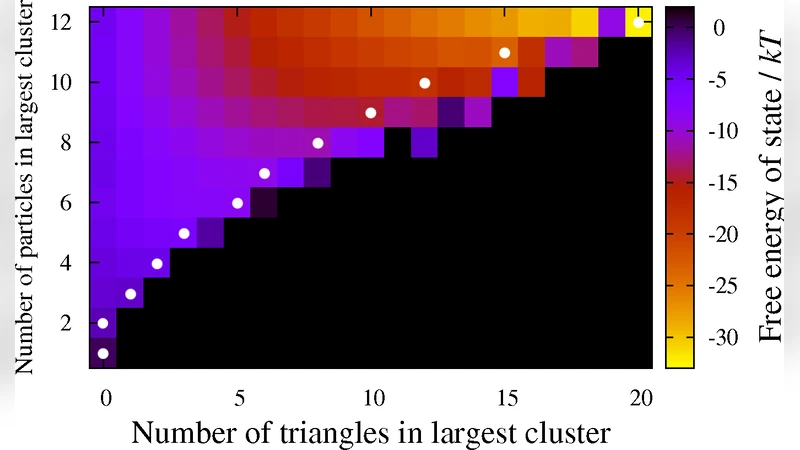
Thermodynamic properties are explored using a combination of Metropolis Monte‑Carlo simulations and umbrella‑sampling techniques to reconstruct free‑energy landscapes across a broad range of (ε, κ) values. The resulting landscapes display a funnel‑like topology: the global minimum corresponds to the target polyhedral shell, while a broad basin of lower‑energy intermediate states guides the system toward that minimum. Crucially, when κ is sufficiently large, the funnel becomes steep and narrow, effectively suppressing the formation of disordered aggregates that would otherwise act as kinetic traps. The authors quantify this effect by measuring the depth of the free‑energy barrier separating the monomeric gas from the nucleated cluster and by evaluating the distribution of structural RMSD values relative to the ideal shell.
Dynamic aspects are investigated by monitoring nucleation rates, growth pathways, and final yields as functions of ε and κ. The study confirms that the nucleation barrier plays a dual role. If the barrier is too low (weak ε or low κ), many nuclei appear simultaneously, rapidly depleting the pool of free monomers and leading to a high incidence of incomplete or malformed shells. Conversely, an excessively high barrier (strong ε combined with very high κ) prevents nucleation altogether, resulting in negligible assembly. Optimal assembly occurs in an intermediate regime where the barrier is high enough to limit the number of concurrent nuclei but low enough to allow nucleation on experimentally relevant timescales. In this regime, monomer supply remains sufficient throughout the growth phase, and the system consistently reaches the target structure with high fidelity.
The authors also examine the specificity of the interactions. By varying κ while keeping ε constant, they demonstrate that a moderate torsional stiffness is essential to discriminate against off‑target binding modes. When κ falls below a critical threshold, the system frequently produces malformed polyhedra, elongated aggregates, or mixed‑symmetry clusters. Above this threshold, the correct angular registry is enforced, and misassembly is dramatically reduced. This finding underscores the importance of rotational specificity in addition to directional attraction for achieving error‑free self‑assembly.
To test the generality of the model, several target geometries are simulated, including icosahedral, dodecahedral, and cubic shells, each with a different number of subunits. In all cases, the same qualitative trends hold: a well‑defined free‑energy funnel, an optimal nucleation barrier, and a requirement for sufficient torsional specificity. The authors further map the simulation parameters onto physical units by calibrating ε and κ against experimentally measured binding free energies and angular tolerances of real viral capsid proteins. When this scaling is applied, the predicted assembly yields and kinetic profiles align closely with published in‑vitro capsid assembly data, providing strong validation of the model’s relevance to real biological systems.
Overall, the work delivers a unified picture of how protein‑like directional and torsional interactions cooperate to produce robust, monodisperse self‑assembly. The key insights are: (1) a moderate free‑energy barrier to nucleation prevents premature monomer depletion; (2) torsional specificity eliminates disordered aggregates and misassembled structures; (3) the resulting free‑energy landscape is funnel‑shaped, ensuring kinetic accessibility of the target shell; and (4) the model’s parameters can be tuned to match real protein systems, making it a valuable tool for both understanding viral capsid formation and guiding the design of synthetic nanocontainers for drug delivery, nanoreactors, or programmable materials.
Comments & Academic Discussion
Loading comments...
Leave a Comment