Resampling Residuals: Robust Estimators of Error and Fit for Evolutionary Trees and Phylogenomics
Phylogenomics, even more so than traditional phylogenetics, needs to represent the uncertainty in evolutionary trees due to systematic error. Here we illustrate the analysis of genome-scale alignments of yeast, using robust measures of the additivity of the fit of distances to tree when using flexi Weighted Least Squares. A variety of DNA and protein distances are used. We explore the nature of the residuals, standardize them, and then create replicate data sets by resampling these residuals. Under the model, the results are shown to be very similar to the conventional sequence bootstrap. With real data they show up uncertainty in the tree that is either due to underestimating the stochastic error (hence massively overestimating the effective sequence length) and/or systematic error. The methods are extended to the very fast BME criterion with similarly promising results.
💡 Research Summary
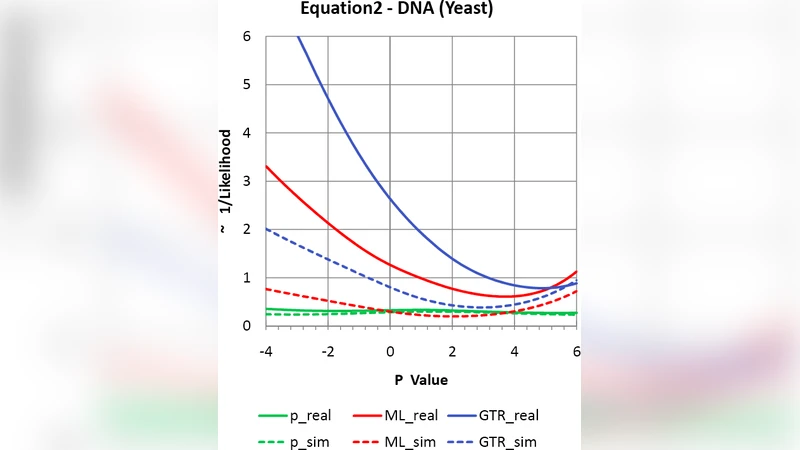
The paper introduces a robust statistical framework for assessing the fit of distance‑based phylogenetic trees and for quantifying uncertainty in large‑scale phylogenomics. Central to the approach is “flexi Weighted Least Squares” (flexi‑WLS), a variant of weighted least‑squares that assigns a dynamic weight to each pairwise distance based on an estimate of its variance. This mitigates the classic problem of heteroscedasticity in distance data, allowing distances derived from different substitution models (e.g., Jukes‑Cantor, Kimura 2‑parameter, Dayhoff) to be combined without bias.
Fit is measured by the additivity residual, the difference between observed and tree‑predicted distances, which is then standardized by the expected standard error of each distance. The standardized residuals provide a scale‑free diagnostic of how well the chosen model explains each entry of the distance matrix.
The novel contribution is “residual resampling”: the standardized residuals are randomly permuted and added back to the tree‑predicted distances to generate a synthetic distance matrix. Flexi‑WLS is re‑applied to each synthetic matrix, producing a replicate tree. Repeating this process thousands of times yields a distribution of topologies and branch lengths analogous to a conventional sequence bootstrap, but derived entirely from the residual structure rather than from resampling the original alignment.
Simulation studies where the model is correctly specified show that residual resampling reproduces bootstrap support values almost identically, confirming that it captures stochastic sampling error accurately. When applied to a genome‑scale alignment of Saccharomyces species, the method reveals substantially lower support for several deep nodes compared with the bootstrap. The authors interpret this discrepancy as evidence of (i) an over‑estimation of the effective sequence length—because the observed variance exceeds the model‑based expectation—and (ii) systematic model misspecification (e.g., rate heterogeneity, inappropriate substitution matrices). High‑variance residuals pinpoint the portions of the data most responsible for these systematic errors, offering a diagnostic that the ordinary bootstrap lacks.
The authors also extend the approach to the Balanced Minimum Evolution (BME) criterion, which is computationally much faster because it directly optimizes a distance‑based objective without iterative tree searches. By applying residual resampling to BME‑derived trees, they obtain confidence measures that closely match those from flexi‑WLS, demonstrating that the technique can be paired with ultra‑fast tree‑building methods without sacrificing statistical rigor.
In summary, the study provides four key advances: (1) a flexible weighted‑least‑squares estimator that accommodates heterogeneous distance variances; (2) a residual‑based resampling scheme that yields bootstrap‑like confidence intervals while being computationally efficient; (3) a means to separate stochastic sampling error from systematic model error, thereby exposing hidden sources of uncertainty in phylogenomic analyses; and (4) validation that the method works equally well with the fast BME algorithm. These contributions make residual resampling a practical alternative to traditional bootstrapping for large phylogenomic data sets, where the latter can be prohibitively expensive and may underestimate true uncertainty.
Comments & Academic Discussion
Loading comments...
Leave a Comment