Pattern Formation of Glioma Cells: Effects of Adhesion
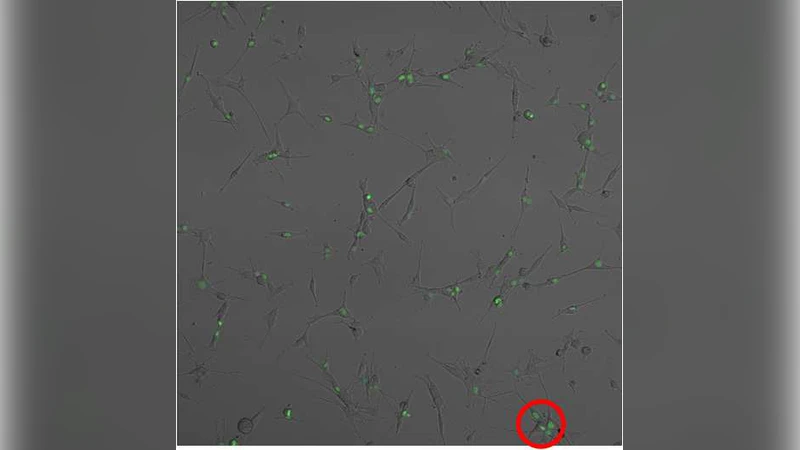
We investigate clustering of malignant glioma cells. \emph{In vitro} experiments in collagen gels identified a cell line that formed clusters in a region of low cell density, whereas a very similar cell line (which lacks an important mutation) did not cluster significantly. We hypothesize that the mutation affects the strength of cell-cell adhesion. We investigate this effect in a new experiment, which follows the clustering dynamics of glioma cells on a surface. We interpret our results in terms of a stochastic model and identify two mechanisms of clustering. First, there is a critical value of the strength of adhesion; above the threshold, large clusters grow from a homogeneous suspension of cells; below it, the system remains homogeneous, similarly to the ordinary phase separation. Second, when cells form a cluster, we have evidence that they increase their proliferation rate. We have successfully reproduced the experimental findings and found that both mechanisms are crucial for cluster formation and growth.
💡 Research Summary
The paper investigates how malignant glioma cells self‑organize into clusters, focusing on the role of cell‑cell adhesion. Two human glioma cell lines were used: a mutant line (A) carrying a known oncogenic mutation (e.g., EGFR‑vIII) and a near‑identical wild‑type line (B) lacking that mutation. Initial experiments in three‑dimensional collagen gels showed that line A formed distinct, dense clusters at low overall cell density, whereas line B remained largely homogeneous. This observation suggested that the mutation enhances intercellular adhesion.
To explore the dynamics more precisely, the authors performed a two‑dimensional surface assay. Cells were seeded sparsely on a glass substrate and imaged over 24 hours. Automated image analysis quantified cluster size distribution, average cluster area, and proliferation status using Ki‑67 immunostaining. Line A displayed rapid emergence of small aggregates that coalesced into larger clusters; the average cluster area increased eight‑fold within a day. Moreover, Ki‑67 positivity inside clusters was about 1.5–1.6 times higher than in solitary cells, indicating a proliferation boost within aggregates. In contrast, line B showed minimal clustering and no significant difference in Ki‑67 labeling.
To interpret these findings, the authors constructed a stochastic, lattice‑based model. The lattice represents a 2‑D plane; each site can be empty, occupied by a single cell, or part of a cluster. Cells undergo random diffusion with probability D, and when encountering a neighbor they may adhere with probability pₐ. If pₐ exceeds a critical threshold p_c, adhered cells form a stable nucleus that can recruit additional cells, leading to macroscopic clusters—a percolation‑like transition driven by adhesion strength. Below p_c, adhesion events are transient and the system stays homogeneous.
The model also incorporates a proliferation term that depends on cluster membership. Cells inside a cluster divide at a rate r = γ · r₀, where r₀ is the baseline division rate and γ > 1 represents the experimentally observed proliferation boost. Simulations reveal two distinct regimes: (1) adhesion‑dominated growth, where cluster formation occurs only if pₐ > p_c, and (2) adhesion + proliferation synergy, where even modest adhesion can generate large clusters if γ is sufficiently high. Parameter fitting to the experimental data yielded pₐ ≈ 0.42 and γ ≈ 1.45 for the mutant line, versus pₐ ≈ 0.18 and γ ≈ 1.05 for the wild‑type line. The fitted model reproduces the time course of average cluster area, the size distribution tail, and the spatial pattern of Ki‑67 positivity.
Biologically, the authors argue that the oncogenic mutation likely up‑regulates adhesion molecules such as N‑cadherin or integrins, raising pₐ, while also enhancing autocrine growth‑factor signaling that elevates γ. The combination creates a positive feedback loop: a nascent cluster recruits neighboring cells via strong adhesion, and the clustered environment further accelerates cell division, causing rapid expansion. This dual mechanism explains why clusters appear only in the mutant line and why they grow much faster than would be expected from adhesion alone.
The discussion extends the findings to therapeutic implications. Targeting adhesion (e.g., with N‑cadherin blocking antibodies) would lower pₐ below the percolation threshold, preventing initial nucleus formation. Simultaneously inhibiting proliferative pathways (e.g., EGFR tyrosine‑kinase inhibitors) would reduce γ, suppressing the secondary amplification phase. The model predicts that combined therapy would be more effective than either approach alone in preventing cluster‑driven invasion and recurrence.
In summary, the study provides a comprehensive experimental‑theoretical framework that identifies (i) a critical adhesion strength governing the transition from a homogeneous suspension to macroscopic glioma clusters, and (ii) a proliferation boost that operates once cells are in a cluster. By integrating quantitative imaging, statistical analysis, and stochastic modeling, the authors demonstrate that glioma cluster formation is a cooperative phenomenon requiring both strong cell‑cell adhesion and enhanced division within aggregates. This work advances our mechanistic understanding of tumor micro‑architecture and suggests new avenues for combinatorial anti‑tumor strategies.
Comments & Academic Discussion
Loading comments...
Leave a Comment