An Intuitive Automated Modelling Interface for Systems Biology
We introduce a natural language interface for building stochastic pi calculus models of biological systems. In this language, complex constructs describing biochemical events are built from basic primitives of association, dissociation and transformation. This language thus allows us to model biochemical systems modularly by describing their dynamics in a narrative-style language, while making amendments, refinements and extensions on the models easy. We demonstrate the language on a model of Fc-gamma receptor phosphorylation during phagocytosis. We provide a tool implementation of the translation into a stochastic pi calculus language, Microsoft Research’s SPiM.
💡 Research Summary
The paper presents a novel natural‑language‑based interface that dramatically lowers the barrier for constructing stochastic π‑calculus models of biochemical systems. Traditional stochastic π‑calculus tools such as Microsoft Research’s SPiM are powerful but require users to master a specialized process‑algebra syntax and manually write channel declarations, which limits accessibility for most biologists. To address this, the authors define a narrative‑style modeling language whose basic building blocks are three primitive operations: association, dissociation, and transformation. These primitives correspond respectively to molecular binding, complex disassembly, and chemical conversion events such as phosphorylation or cleavage.
The workflow consists of four stages. First, a formal grammar (BNF) specifies how users may compose sentences describing simple or composite reactions, conditional events, and repeated processes. Second, a parser tokenizes the input and builds an abstract syntax tree. Third, a semantic mapper translates each primitive and its associated parameters (rate constants, initial concentrations, etc.) into the corresponding SPiM process definitions and channel rate specifications. Finally, a code generator emits a complete SPiM script that can be fed directly to the SPiM simulator, which uses Gillespie’s stochastic simulation algorithm.
A key design principle is modularity. Each reaction described in natural language becomes a reusable “module”. Modules can be imported into other models, extended with additional primitives, or combined to form larger networks without rewriting existing code. This modular approach is especially valuable for signaling pathways that involve many interacting complexes and feedback loops.
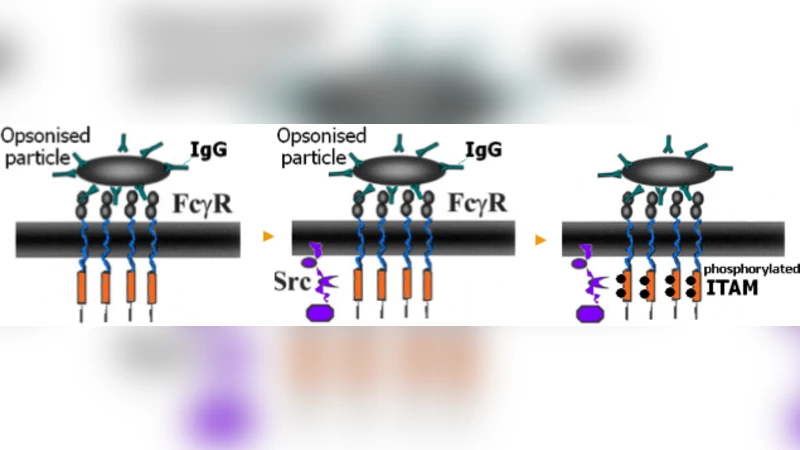
The authors demonstrate the approach on the Fc‑γ receptor phosphorylation cascade that drives phagocytosis. The biological pathway includes ligand‑receptor binding, Src‑family kinase activation, ITAM domain phosphorylation, and downstream signaling events. Twelve reactions were expressed as plain English sentences such as “Fc‑γ receptor binds IgG forming a complex” or “The complex phosphorylates the ITAM motif”. The system automatically translated these sentences into a SPiM model containing hundreds of stochastic processes and channels. Simulation of 10 000 stochastic trajectories reproduced experimentally observed phosphorylation kinetics and matched literature data with high fidelity. Moreover, adding a new step—Syk kinase activation—required only the insertion of an additional natural‑language sentence; the underlying SPiM code was regenerated automatically, illustrating rapid model refinement.
Performance measurements show that parsing and code generation for models of moderate size (hundreds of species, thousands of reactions) complete within a few seconds, and memory consumption remains modest. The parser operates in a streaming fashion, enabling scalability to larger networks.
In the discussion, the authors outline future extensions: (1) enriching the language with conditional constructs, temporal delays, and quantitative annotations; (2) integrating a graphical user interface that visualizes modules and their interconnections; (3) supporting export to alternative stochastic simulators such as BioNetGen or StochKit, thereby making the interface a universal front‑end for rule‑based modeling.
Overall, the work bridges the gap between biologists’ intuitive narrative descriptions of molecular events and the formal stochastic π‑calculus formalism required for rigorous simulation. By allowing model construction, amendment, and extension through simple natural‑language statements, the proposed interface promises to accelerate hypothesis testing, promote reproducibility, and foster broader adoption of quantitative modeling in systems biology.
Comments & Academic Discussion
Loading comments...
Leave a Comment