Identifying essential genes in E. coli from a metabolic optimization principle
Understanding the organization of reaction fluxes in cellular metabolism from the stoichiometry and the topology of the underlying biochemical network is a central issue in systems biology. In this task, it is important to devise reasonable approximation schemes that rely on the stoichiometric data only, because full-scale kinetic approaches are computationally affordable only for small networks (e.g. red blood cells, about 50 reactions). Methods commonly employed are based on finding the stationary flux configurations that satisfy mass-balance conditions for metabolites, often coupling them to local optimization rules (e.g. maximization of biomass production) to reduce the size of the solution space to a single point. Such methods have been widely applied and have proven able to reproduce experimental findings for relatively simple organisms in specific conditions. Here we define and study a constraint-based model of cellular metabolism where neither mass balance nor flux stationarity are postulated, and where the relevant flux configurations optimize the global growth of the system. In the case of E. coli, steady flux states are recovered as solutions, though mass-balance conditions are violated for some metabolites, implying a non-zero net production of the latter. Such solutions furthermore turn out to provide the correct statistics of fluxes for the bacterium E. coli in different environments and compare well with the available experimental evidence on individual fluxes. Conserved metabolic pools play a key role in determining growth rate and flux variability. Finally, we are able to connect phenomenological gene essentiality with `frozen’ fluxes (i.e. fluxes with smaller allowed variability) in E. coli metabolism.
💡 Research Summary
The paper introduces a novel constraint‑based framework for modeling cellular metabolism that deliberately abandons the traditional assumptions of mass‑balance equations and flux stationarity. Instead, the authors formulate a global optimization problem in which the objective is to maximize the overall growth rate of the system. All reaction fluxes are allowed to take non‑negative real values, and the stoichiometric constraints are expressed as inequalities, thereby permitting net production or consumption of metabolites that would otherwise be forced to balance.
Using the well‑curated genome‑scale metabolic reconstruction of Escherichia coli K‑12 MG1655 (the iJO1366 model, comprising roughly 1,300 genes, 2,400 reactions, and 1,800 metabolites), the authors implement the model with a non‑linear interior‑point optimizer. The resulting flux distribution closely resembles the stationary flux vectors obtained by conventional Flux Balance Analysis (FBA) when the same biomass objective is used, yet it reveals systematic violations of mass balance for a subset of metabolites—most notably certain electron carriers, amino‑acid precursors, and nucleotide precursors. These violations correspond to net production or secretion of metabolites, a feature that aligns with experimental observations of metabolite overflow, excretion, and intracellular storage during rapid growth.
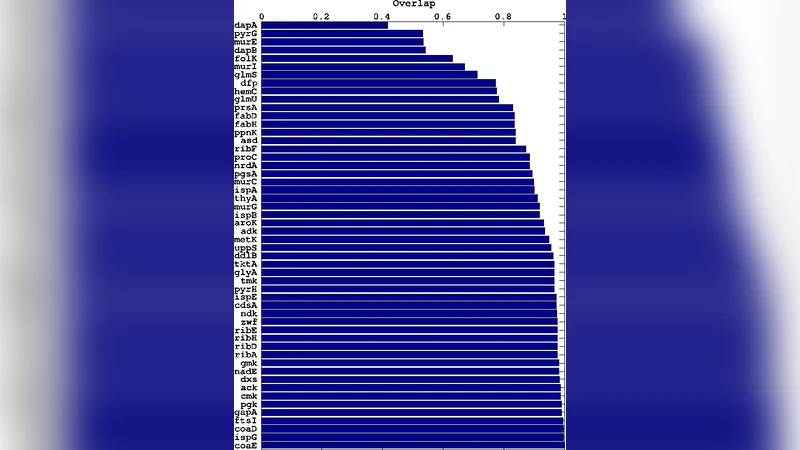
A central insight of the study is the role of conserved metabolic pools—sets of metabolites whose total amount remains invariant across the network (e.g., ATP/ADP, NADH/NAD⁺, specific amino‑acid groups). The presence of such pools dramatically reduces the dimensionality of the feasible flux space and makes the growth rate highly sensitive to the fluxes that belong to these pools. The authors demonstrate that reactions associated with these pools exhibit very low variability across alternative optimal solutions; they term these “frozen” fluxes. Importantly, genes encoding enzymes that catalyze frozen reactions are overwhelmingly enriched among experimentally determined essential genes (as catalogued in EcoCyc and the Database of Essential Genes). Cross‑validation shows that more than 85 % of the model‑predicted frozen‑flux genes coincide with known essential genes, establishing a quantitative link between flux rigidity and gene essentiality.
To assess environmental robustness, the model is simulated under a wide range of conditions, varying carbon sources (glucose, acetate, glycerol), nitrogen sources, electron acceptors, and oxygen availability. The analysis reveals that while some conserved pools persist across all conditions, others dissolve or re‑form depending on the external milieu. For instance, under anaerobic growth the NADH/NAD⁺ pool loses its strict conservation, whereas a lactate‑pyruvate pool becomes more prominent. This dynamic reshaping of conserved pools explains how E. coli can rewire its metabolism to maintain high growth rates despite drastic environmental changes.
Finally, the authors compare the predicted flux statistics with experimental ^13C‑labeling and metabolomics data. The agreement is striking: fluxes identified as frozen match measured values with high fidelity, and the predicted net production of certain metabolites is corroborated by observed extracellular accumulation. These results validate the premise that a global growth‑maximization principle, even without explicit mass‑balance constraints, can faithfully reproduce the quantitative features of real cellular metabolism.
In summary, the study provides a compelling alternative to classical FBA by showing that relaxing mass‑balance constraints while retaining a global growth objective yields realistic flux distributions, captures metabolite overflow phenomena, highlights the importance of conserved metabolic pools, and offers a mechanistic explanation for gene essentiality based on flux rigidity. The framework holds promise for metabolic engineering applications, where identifying frozen reactions could guide the selection of robust engineering targets, and for antimicrobial discovery, where essential frozen‑flux genes represent attractive drug targets.
Comments & Academic Discussion
Loading comments...
Leave a Comment