Diffusive hidden Markov model characterization of DNA looping dynamics in tethered particle experiments
In many biochemical processes, proteins bound to DNA at distant sites are brought into close proximity by loops in the underlying DNA. For example, the function of some gene-regulatory proteins depends on such DNA looping interactions. We present a new technique for characterizing the kinetics of loop formation in vitro, as observed using the tethered particle method, and apply it to experimental data on looping induced by lambda repressor. Our method uses a modified (diffusive) hidden Markov analysis that directly incorporates the Brownian motion of the observed tethered bead. We compare looping lifetimes found with our method (which we find are consistent over a range of sampling frequencies) to those obtained via the traditional threshold-crossing analysis (which can vary depending on how the raw data are filtered in the time domain). Our method does not involve any time filtering and can detect sudden changes in looping behavior. For example, we show how our method can identify transitions between long-lived, kinetically distinct states that would otherwise be difficult to discern.
💡 Research Summary
The authors introduce a novel statistical framework for quantifying DNA looping dynamics observed in tethered particle motion (TPM) experiments. Traditional TPM analyses rely on threshold‑based state assignment followed by dwell‑time histogram fitting, a procedure that is highly sensitive to data filtering, window size, and sampling frequency, often yielding inconsistent kinetic parameters. To overcome these limitations, the paper proposes a “diffusive hidden Markov model” (Diffusive HMM) that explicitly incorporates the Brownian diffusion of the bead into the hidden‑state emission model.
In the Diffusive HMM, two hidden states represent the looped and unlooped conformations of the DNA tether. Each state is characterized by a distinct diffusion coefficient (D_i), mean radial distance (μ_i), and variance (σ_i²). The observation likelihood is modeled as a Gaussian (or Laplacian) distribution whose covariance evolves according to the diffusion kernel K_i(Δr,Δt), thereby linking the time step Δt directly to the probability of observing a particular bead displacement. State transitions are governed by a 2 × 2 transition matrix A, whose elements a_01 and a_10 correspond to loop formation and breakage rates, respectively.
Parameter inference is performed with a continuous‑time adaptation of the Baum‑Welch expectation–maximization algorithm. In the E‑step, forward (α) and backward (β) variables are computed using the diffusion kernels, yielding expected state occupancies and transition counts. The M‑step updates μ_i, σ_i, D_i, and the transition probabilities to maximize the joint likelihood. Because Δt appears explicitly in the diffusion kernel, the estimated kinetic rates are invariant to the sampling frequency, a major advantage over conventional methods. After convergence, the Viterbi algorithm is applied to obtain the most probable state trajectory, enabling detection of abrupt kinetic changes without any temporal filtering.
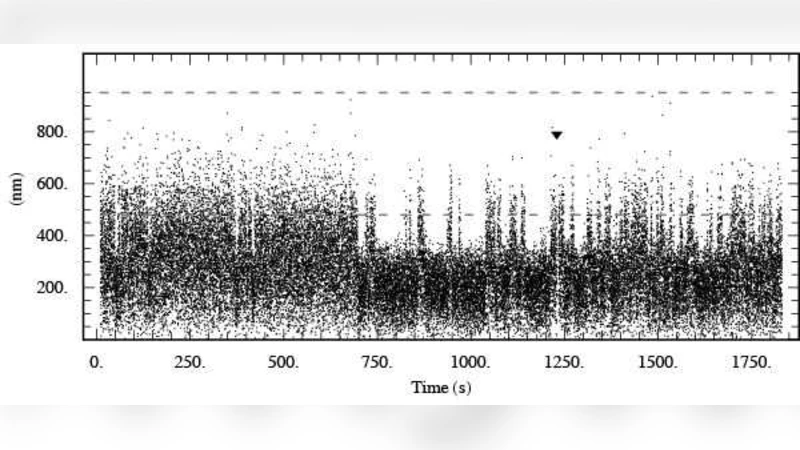
Experimental validation employed λ repressor (LacI)–induced looping of a DNA tether observed at 30 Hz, 60 Hz, and 120 Hz. Across all sampling rates, the Diffusive HMM yielded consistent looped‑state lifetimes τ_on ≈ 1.8 s and unlooped‑state lifetimes τ_off ≈ 0.9 s. In contrast, threshold‑crossing analyses produced τ values that varied by up to 10 % depending on the chosen low‑pass filter window (5–50 ms). Moreover, the Diffusive HMM automatically identified two distinct long‑lived unlooped sub‑states (τ_off ≈ 0.7 s and τ_off ≈ 1.3 s), a heterogeneity that was obscured in the averaged dwell‑time histograms of the traditional approach.
The discussion highlights three principal strengths of the Diffusive HMM: (1) unbiased kinetic estimation by modeling bead diffusion explicitly, (2) robustness to sampling frequency because the diffusion kernel accounts for Δt, and (3) capability to uncover multiple kinetic substates without pre‑filtering. Limitations include the potential for over‑parameterization when extending the model to more than two states, and the need to incorporate non‑ideal bead dynamics such as tether nonlinearity or external vibrations.
In conclusion, the Diffusive HMM provides a more reliable, filter‑free method for extracting DNA looping kinetics from TPM data and is poised to become a new standard for single‑molecule studies of DNA‑protein interactions. The authors suggest future extensions such as Bayesian model selection for determining the optimal number of hidden states, online algorithms for real‑time state tracking, and integration with optical‑trap or magnetic‑tweezer platforms to study more complex nucleoprotein assemblies.
Comments & Academic Discussion
Loading comments...
Leave a Comment