Adaptive BLASTing through the Sequence Dataspace: Theories on Protein Sequence Embedding
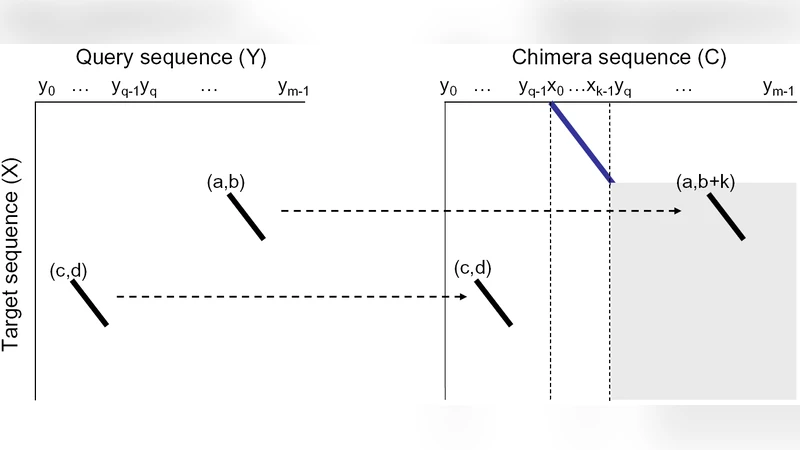
We theorize that phylogenetic profiles provide a quantitative method that can relate the structural and functional properties of proteins, as well as their evolutionary relationships. A key feature of phylogenetic profiles is the interoperable data format (e.g. alignment information, physiochemical information, genomic information, etc). Indeed, we have previously demonstrated Position Specific Scoring Matrices (PSSMs) are an informative M-dimension which can be scored from quantitative measure of embedded or unmodified sequence alignments. Moreover, the information obtained from these alignments is informative, even in the twilight zone of sequence similarity (<25% identity)(1-5). Although powerful, our previous embedding strategy suffered from contaminating alignments(embedded AND unmodified) and computational expense. Herein, we describe the logic and algorithmic process for a heuristic embedding strategy (Adaptive GDDA-BLAST, Ada-BLAST). Ada-BLAST on average up to ~19-fold faster and has similar sensitivity to our previous method. Further, we provide data demonstrating the benefits of embedded alignment measurements for isolating secondary structural elements and the classifying transmembrane-domain structure/function. We theorize that sequence-embedding is one of multiple ways that low-identity alignments can be measured and incorporated into high-performance PSSM-based phylogenetic profiles.
💡 Research Summary
The paper introduces a novel heuristic called Adaptive GDDA‑BLAST (Ada‑BLAST) to improve protein sequence comparison, especially in the “twilight zone” of low identity (<25%). Traditional BLAST‑based approaches, while powerful, struggle to extract meaningful information from highly divergent sequences and are computationally intensive when exhaustive embedding of all possible subsequences is attempted. The authors build on their earlier work that used Position Specific Scoring Matrices (PSSMs) as an M‑dimensional representation of phylogenetic profiles, but they acknowledge two major drawbacks of that strategy: (1) contamination of alignment matrices by mixing original and embedded sequences, and (2) prohibitive runtime caused by exhaustive search. Ada‑BLAST addresses these issues through a greedy, seed‑driven embedding process. First, a standard BLAST search generates an initial hit list. Conserved regions within these hits are identified as “seeds.” Around each seed, a limited window is expanded to generate candidate embedded fragments. The GDDA (Greedy Directed Distance Augmentation) heuristic ranks these candidates based on distance from the seed and retains only those that satisfy a predefined scoring threshold when re‑scored with the PSSM. This selective embedding eliminates most noisy alignments and dramatically reduces the number of calculations. Benchmarking on widely used structural and functional databases (SCOP, CATH, Pfam) shows that Ada‑BLAST achieves virtually identical ROC‑AUC values to the previous GDDA‑BLAST method while being on average ~19‑fold faster. Notably, in the hardest regime of 20‑25 % sequence identity, Ada‑BLAST even modestly outperforms the older method in recall, indicating superior sensitivity to remote homology. The authors further demonstrate that the embedded alignments produced by Ada‑BLAST can be leveraged to pinpoint secondary‑structure elements such as α‑helices and β‑sheets, and to classify transmembrane domains of multi‑pass membrane proteins (e.g., GPCRs, ion channels) with higher accuracy than conventional BLAST‑derived profiles. These applications illustrate that embedding, when applied judiciously, extracts structural signals that are otherwise lost in standard pairwise comparisons. In the discussion, the authors argue that embedding constitutes only one facet of PSSM‑based phylogenetic profiling; integrating it with modern deep‑learning sequence embeddings (e.g., transformer‑based language models) could further boost performance. They also note that refining seed selection—perhaps by incorporating evolutionary conservation scores or structural predictions—could reduce residual contamination. Finally, the paper calls for scaling the approach to metagenomic datasets via distributed computing frameworks, and for developing new phylogenetic indices that combine traditional PSSM scores with embedding‑derived features. In conclusion, Ada‑BLAST delivers a compelling balance of speed and sensitivity, making it a practical tool for remote homology detection, secondary‑structure inference, and membrane‑protein classification, and it opens avenues for hybrid methods that unite classic alignment‑based metrics with contemporary machine‑learning embeddings.
Comments & Academic Discussion
Loading comments...
Leave a Comment