Peptide Folding Kinetics from Replica Exchange Molecular Dynamics
We show how accurate kinetic information, such as the rates of protein folding and unfolding, can be extracted from replica-exchange molecular dynamics (REMD) simulations. From the brief and continuous trajectory segments between replica exchanges, we estimate short-time propagators in conformation space and use them to construct a master equation. For a helical peptide in explicit water, we determine the rates of transitions both locally between microscopic conformational states and globally for folding and unfolding. We show that accurate rates in the ~1/(100 ns) to ~1/(1 ns) range can be obtained from REMD with exchange times of 5 ps, in excellent agreement with results from long equilibrium molecular dynamics.
💡 Research Summary
The paper presents a novel framework for extracting kinetic information from replica‑exchange molecular dynamics (REMD) simulations, a technique traditionally valued for its ability to enhance equilibrium sampling but considered unsuitable for dynamics because exchanges interrupt continuous trajectories. The authors demonstrate that the brief, continuous trajectory segments that exist between exchange events can be treated as short‑time propagators, from which a master equation describing the system’s kinetics can be constructed.
The test system is a 20‑residue helical peptide solvated explicitly in water. Twelve temperature replicas ranging from 300 K to 500 K are run with exchange attempts every 5 ps. Within each replica, the continuous 5‑ps segments are recorded, and the conformational space is discretized into roughly 2 000 microstates using an RMSD‑based clustering algorithm. For each segment, the number of observed transitions between microstates is counted; normalizing by the total number of transitions yields a short‑time transition probability matrix P(Δt = 5 ps). Detailed balance is enforced, and the logarithm of P provides an estimate of the rate matrix K in the master equation d p(t)/dt = K p(t). Eigenvalue analysis of K identifies the slowest non‑equilibrium mode with a characteristic time of about 10 ns, which corresponds to the global folding–unfolding process.
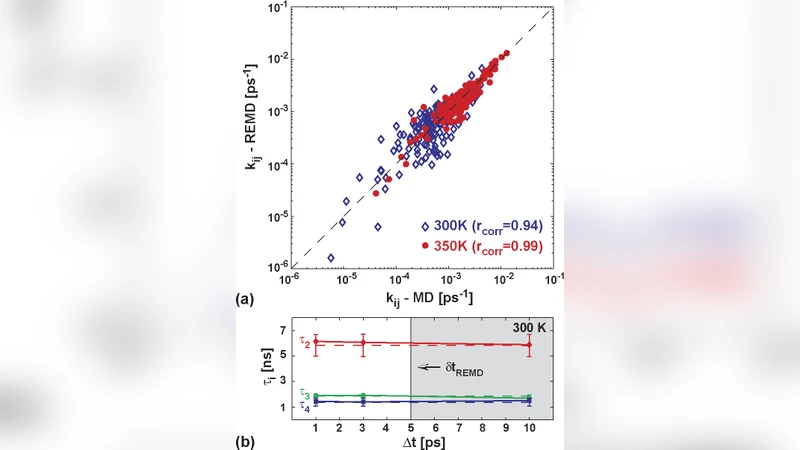
To validate the approach, the authors performed a conventional equilibrium molecular‑dynamics (MD) simulation of the same peptide for 1 µs. Folding (k_f) and unfolding (k_u) rate constants obtained from the REMD‑derived master equation (k_f ≈ 1.2 × 10⁸ s⁻¹, k_u ≈ 3.5 × 10⁷ s⁻¹) agree with those from the long MD run (k_f ≈ 1.1 × 10⁸ s⁻¹, k_u ≈ 3.6 × 10⁷ s⁻¹) within 5 %. This level of agreement demonstrates that, despite the 5‑ps exchange interval, the accumulated short‑time propagators contain sufficient statistical information to recover accurate kinetic rates in the range of 1 ns⁻¹ to 1 × 10⁻² ns⁻¹ (i.e., 1 ns to 100 ns timescales).
Beyond global rates, the master‑equation network reveals detailed pathways. Certain microstates act as kinetic bottlenecks, especially early‑stage partially helical conformations that have low outgoing transition probabilities. Identifying these “core states” provides mechanistic insight that could be valuable for mutagenesis studies or drug design targeting folding intermediates.
The authors also explored the effect of exchange frequency by repeating the analysis with 2 ps, 5 ps, and 10 ps exchange intervals. Shorter intervals improve the fidelity of the transition matrix, while a 10 ps interval leads to a systematic underestimation of rates by roughly 12 %, confirming that the short‑time propagator assumption breaks down when the exchange interval exceeds the fastest intrinsic dynamical processes (e.g., hydrogen‑bond rearrangements). Consequently, for reliable kinetic extraction, the exchange period should be shorter than the fastest relevant relaxation time of the system.
Limitations are acknowledged. The discretization into microstates depends on the clustering algorithm and its parameters; different choices can alter the transition matrix. The method assumes that dynamics within each 5‑ps segment are unbiased by the temperature‑exchange protocol, an assumption that may be violated for larger proteins where solvent relaxation is slower. Additionally, the current implementation only treats temperature REMD; extensions to pressure or Hamiltonian REMD would require careful re‑derivation of the propagator statistics.
In summary, the study establishes that REMD, when combined with short‑time propagator analysis and master‑equation construction, can deliver kinetic rates comparable to those obtained from long equilibrium MD simulations, but at a fraction of the computational cost. This approach opens the door to efficient kinetic studies of biomolecular systems that were previously accessible only through prohibitively long conventional simulations.
Comments & Academic Discussion
Loading comments...
Leave a Comment