The limited role of non-native contacts in folding pathways of a lattice protein
Models of protein energetics which neglect interactions between amino acids that are not adjacent in the native state, such as the Go model, encode or underlie many influential ideas on protein folding. Implicit in this simplification is a crucial assumption that has never been critically evaluated in a broad context: Detailed mechanisms of protein folding are not biased by non-native contacts, typically imagined as a consequence of sequence design and/or topology. Here we present, using computer simulations of a well-studied lattice heteropolymer model, the first systematic test of this oft-assumed correspondence over the statistically significant range of hundreds of thousands of amino acid sequences, and a concomitantly diverse set of folding pathways. Enabled by a novel means of fingerprinting folding trajectories, our study reveals a profound insensitivity of the order in which native contacts accumulate to the omission of non-native interactions. Contrary to conventional thinking, this robustness does not arise from topological restrictions and does not depend on folding rate. We find instead that the crucial factor in discriminating among topological pathways is the heterogeneity of native contact energies. Our results challenge conventional thinking on the relationship between sequence design and free energy landscapes for protein folding, and help justify the widespread use of Go-like models to scrutinize detailed folding mechanisms of real proteins.
💡 Research Summary
This paper provides the first large‑scale systematic test of a central assumption underlying many protein‑folding models, especially Go‑type models that ignore non‑native contacts. Using a classic three‑dimensional lattice heteropolymer (27‑mer HP model), the authors generated on the order of three hundred thousand distinct amino‑acid sequences, each of which folds via a diverse set of pathways. For every sequence two energy functions were employed: (i) a full potential that includes both native and non‑native pairwise interactions, and (ii) a Go‑type potential that retains only native contacts. Monte‑Carlo and molecular‑dynamics simulations were run for each case, producing thousands of folding trajectories.
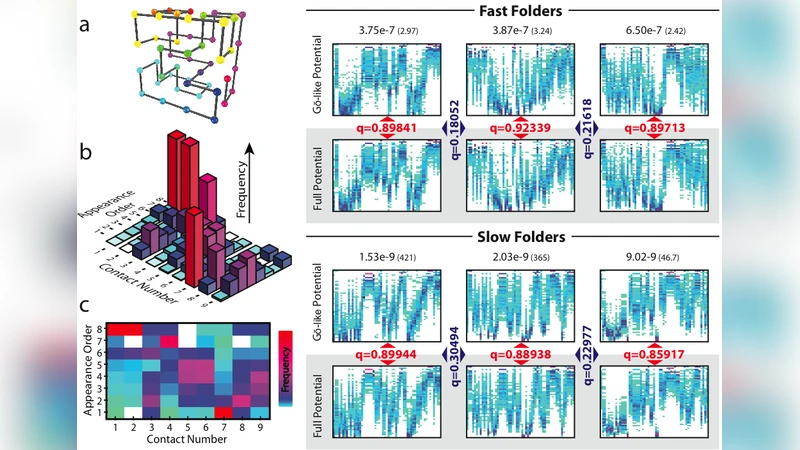
A novel “folding trajectory fingerprint” was introduced to compare pathways quantitatively. Each trajectory was encoded as a binary vector indicating the order in which native contacts become established; the vectors were then compared across the two potentials using edit‑distance metrics and hierarchical clustering. This approach allowed the authors to assess directly whether the omission of non‑native interactions reshapes the sequence of native‑contact formation.
The results are strikingly consistent. Across the massive ensemble of sequences, the similarity between trajectories generated with the full potential and those generated with the Go‑type potential is high (average similarity > 0.85). In other words, the order in which native contacts accumulate is largely insensitive to the presence or absence of non‑native contacts. This insensitivity does not stem from topological constraints: clustering based solely on the native contact network yields virtually identical groups for both potentials. Moreover, the effect is independent of folding speed; fast‑folding and slow‑folding sequences show comparable trajectory overlap.
The decisive factor that differentiates pathways is the heterogeneity of native‑contact energies. When a sequence’s native contacts have a narrow energy distribution, folding pathways are constrained and similar across models. By contrast, sequences with a broad spread of native‑contact energies display distinct ordering: contacts with more favorable (lower) energies tend to form early, steering the pathway. Thus, the variability in native‑contact energetics—not the presence of non‑native interactions—governs the detailed folding route.
These findings overturn the conventional view that sequence design primarily reduces non‑native contacts to smooth the energy landscape. Instead, the study suggests that the design of native‑contact energy heterogeneity is the key to shaping folding mechanisms. Consequently, the authors argue that Go‑type models, despite their simplifications, are justified for probing detailed folding pathways of real proteins because the omitted non‑native contacts do not substantially alter the order of native‑contact formation.
Beyond the specific lattice system, the work introduces a robust methodological framework for fingerprinting folding trajectories, which can be applied to off‑lattice and atomistic simulations. By demonstrating that non‑native interactions have a limited role in dictating pathway topology, the paper provides a strong theoretical foundation for the continued use of simplified, native‑centric models in protein‑folding research, while also highlighting the importance of native‑contact energy heterogeneity as a design principle for both natural and engineered proteins.
Comments & Academic Discussion
Loading comments...
Leave a Comment