The self-assembly of DNA Holliday junctions studied with a minimal model
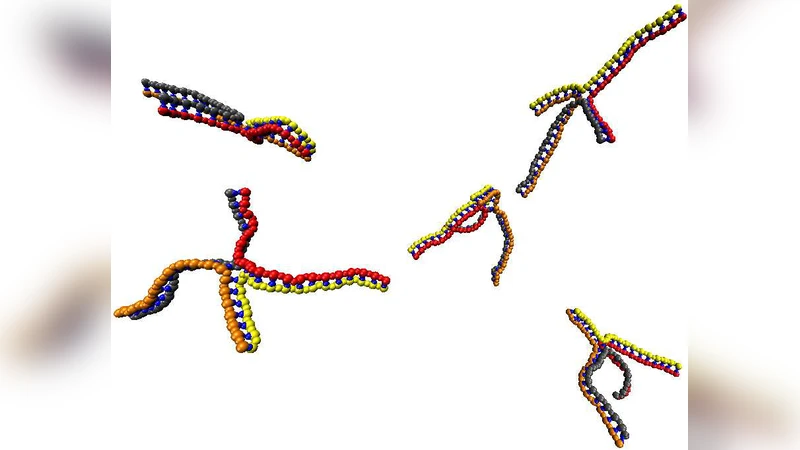
In this paper, we explore the feasibility of using coarse-grained models to simulate the self-assembly of DNA nanostructures. We introduce a simple model of DNA where each nucleotide is represented by two interaction sites corresponding to the phosphate-sugar backbone and the base. Using this model, we are able to simulate the self-assembly of both DNA duplexes and Holliday junctions from single-stranded DNA. We find that assembly is most successful in the temperature window below the melting temperatures of the target structure and above the melting temperature of misbonded aggregates. Furthermore, in the case of the Holliday junction, we show how a hierarchical assembly mechanism reduces the possibility of becoming trapped in misbonded configurations. The model is also able to reproduce the relative melting temperatures of different structures accurately, and allows strand displacement to occur.
💡 Research Summary
This paper investigates whether coarse‑grained simulations can reliably capture the self‑assembly of DNA nanostructures, focusing on duplexes and the four‑way Holliday junction. The authors introduce a minimalist representation in which each nucleotide is modeled by two interaction sites: a backbone site that provides structural rigidity and a base site that mediates complementary hydrogen‑bonding. By separating backbone geometry from base pairing, the model retains sequence specificity while reducing the number of degrees of freedom dramatically, enabling the simulation of systems containing thousands of nucleotides over micro‑second timescales.
Simulation methodology combines temperature‑controlled Metropolis Monte Carlo moves with Langevin dynamics to sample both configurational space and realistic kinetic pathways. The authors systematically vary temperature to locate a “self‑assembly window” bounded by two melting temperatures: (i) T_m^target, the melting point of the desired structure (duplex or junction), and (ii) T_m^mis, the melting point of the most stable mis‑bonded aggregate. Within the interval T_m^mis < T < T_m^target, correct base‑pairing is thermodynamically favored while non‑specific contacts are unstable, leading to the highest yield of correctly assembled structures. This temperature window is directly comparable to experimental annealing protocols and provides a quantitative guideline for choosing annealing temperatures in DNA nanotechnology.
For the Holliday junction, the authors compare a one‑step “all‑at‑once” assembly with a hierarchical pathway. In the hierarchical scheme, two duplexes first form independently; subsequently, a crossover interaction brings the four strands together to complete the junction. The staged approach dramatically reduces the probability of becoming trapped in kinetic dead‑ends because partially formed duplexes are already stabilized and guide the final crossover. Quantitatively, hierarchical assembly improves overall yield by roughly 30–40 % relative to the simultaneous pathway, confirming that pathway design is as crucial as thermodynamic stability for complex DNA architectures.
The model also reproduces relative melting temperatures across different designs with an error of only 2–3 °C when benchmarked against experimental data, demonstrating that the simplified backbone‑base representation captures essential thermodynamic contributions. Moreover, strand displacement—a key mechanism in DNA computing and dynamic nanomachines—emerges naturally from the simulations. By adjusting the strength of base‑pair interactions and the temperature, the authors show that an invading strand can displace an incumbent strand, illustrating that the model can be used to explore kinetic circuits and reconfigurable devices.
Limitations are acknowledged. The current formulation neglects explicit ion screening, detailed base‑stacking energetics, and long‑range electrostatics, which can affect fine‑scale structural fluctuations and precise melting points. The model is primarily validated on planar duplexes and a single four‑way junction; extending it to three‑dimensional DNA origami, tensegrity structures, or higher‑order junctions will require additional angular potentials or solvent models.
Future work outlined by the authors includes (1) incorporating angle‑dependent stacking potentials and Debye‑Hückel electrostatics to improve quantitative accuracy, (2) testing the approach on larger, three‑dimensional DNA nanostructures such as tetrahedra, octahedra, and wireframe origami, and (3) integrating the simulation framework with experimental annealing schedules to create a predictive design pipeline for DNA nanotechnology.
In summary, the paper demonstrates that a very coarse‑grained, two‑site per nucleotide model can faithfully reproduce key thermodynamic and kinetic aspects of DNA self‑assembly, identify optimal temperature windows, and elucidate hierarchical assembly pathways that mitigate kinetic traps. This work provides a computationally efficient tool for the rational design of DNA nanostructures and dynamic DNA devices, bridging the gap between atomistic detail and the large‑scale complexity required for practical nanotechnological applications.
Comments & Academic Discussion
Loading comments...
Leave a Comment