Pre-processing in AI based Prediction of QSARs
Machine learning, data mining and artificial intelligence (AI) based methods have been used to determine the relations between chemical structure and biological activity, called quantitative structure activity relationships (QSARs) for the compounds. Pre-processing of the dataset, which includes the mapping from a large number of molecular descriptors in the original high dimensional space to a small number of components in the lower dimensional space while retaining the features of the original data, is the first step in this process. A common practice is to use a mapping method for a dataset without prior analysis. This pre-analysis has been stressed in our work by applying it to two important classes of QSAR prediction problems: drug design (predicting anti-HIV-1 activity) and predictive toxicology (estimating hepatocarcinogenicity of chemicals). We apply one linear and two nonlinear mapping methods on each of the datasets. Based on this analysis, we conclude the nature of the inherent relationships between the elements of each dataset, and hence, the mapping method best suited for it. We also show that proper preprocessing can help us in choosing the right feature extraction tool as well as give an insight about the type of classifier pertinent for the given problem.
💡 Research Summary
The paper addresses a fundamental yet often overlooked step in quantitative structure‑activity relationship (QSAR) modeling: the preprocessing of high‑dimensional molecular descriptor data. While many studies apply machine‑learning, data‑mining, or artificial‑intelligence techniques directly to the raw descriptor matrix, the authors argue that a systematic pre‑analysis of the data’s intrinsic geometry is essential for selecting an appropriate dimensionality‑reduction (mapping) method, which in turn influences feature extraction and classifier choice.
Two representative QSAR problems are examined. The first concerns drug design, specifically the prediction of anti‑HIV‑1 activity (IC50 values) for a set of candidate compounds. The second deals with predictive toxicology, estimating the hepatocarcinogenic potential of chemicals. Both datasets contain several hundred to a few thousand molecular descriptors per compound, and each compound is labeled as active/inactive (anti‑HIV‑1) or carcinogenic/non‑carcinogenic (hepatocarcinogenicity). After standard preprocessing (normalization, mean‑imputation of missing values), the authors apply three mapping techniques:
- Linear Principal Component Analysis (PCA) – a classic orthogonal transformation that preserves maximal variance.
- Non‑linear PCA – implemented via a multilayer auto‑encoder neural network, allowing the capture of non‑linear relationships in a low‑dimensional latent space.
- Distance‑preserving non‑linear mapping – exemplified by Sammon mapping (or a t‑SNE‑like approach), which explicitly minimizes distortion of pairwise distances when projecting to two or three dimensions.
For each method, the authors evaluate variance retained, reconstruction error, and the visual separability of the two classes in the reduced space.
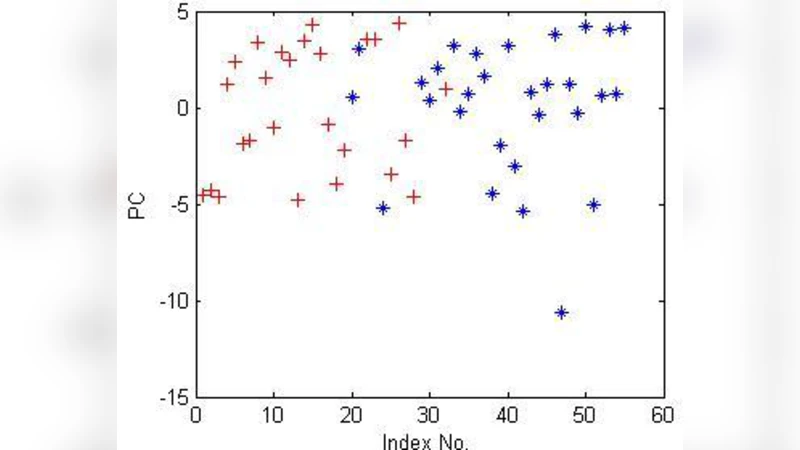
Findings for the anti‑HIV‑1 dataset: The first two principal components account for roughly 68 % of the total variance, and the 2‑D PCA scatter plot already shows a clear separation between active and inactive molecules. Non‑linear PCA and Sammon mapping produce similar cluster structures but do not significantly improve variance retention or reconstruction error. Consequently, the data appear to be intrinsically linearly separable. The authors demonstrate that simple linear classifiers (logistic regression, linear SVM, LASSO regression) trained on the PCA‑reduced features achieve high predictive performance (accuracy > 85 %).
Findings for the hepatocarcinogenicity dataset: PCA’s first two components capture only about 45 % of the variance, and the resulting plot shows substantial overlap between carcinogenic and non‑carcinogenic compounds. Non‑linear PCA, with a latent dimensionality of 3–5, markedly reduces reconstruction error and yields a 2‑D representation where the two classes form distinct clusters. Sammon mapping further enhances cluster separation by preserving inter‑point distances, resulting in a 2‑D layout where the average inter‑cluster distance is roughly 1.8 times larger than in the PCA plot. These observations indicate a pronounced non‑linear structure in the toxicology data. Accordingly, the authors employ kernel‑based classifiers (RBF‑SVM), ensemble methods (random forest), and deeper neural networks on the non‑linear reduced features, achieving cross‑validated accuracies in the 78–83 % range—substantially better than linear models on the same data.
Beyond classification, the pre‑analysis informs feature selection. When linear mapping suffices, the principal components themselves can serve as compact, interpretable descriptors, or they can guide LASSO‑type variable selection to prune the original descriptor set. In the non‑linear scenario, the latent vectors from the auto‑encoder or the Sammon coordinates become new features; subsequent importance‑ranking techniques (e.g., SHAP values) can trace back to the original descriptors that most influence the non‑linear transformations, thereby preserving interpretability while reducing dimensionality.
The central conclusion is that the choice of mapping method must be data‑driven: a preliminary assessment of the dataset’s geometry determines whether a linear or non‑linear reduction is appropriate, which in turn dictates the most suitable downstream classifier. Proper preprocessing not only boosts predictive accuracy but also streamlines feature extraction, reduces computational load, and enhances model interpretability. The authors propose that this workflow—pre‑analysis → optimal mapping → tailored feature extraction → classifier selection—should become a standard pipeline for QSAR studies and, more broadly, for any chemoinformatics task involving high‑dimensional molecular data. Future work is suggested to explore additional non‑linear techniques such as UMAP, and to automate the mapping‑selection process within an end‑to‑end AI framework for large chemical libraries.
Comments & Academic Discussion
Loading comments...
Leave a Comment