Ab-initio Dynamics of Rare Thermally Activated Reactions
We introduce a framework to investigate ab-initio the dynamics of rare thermally activated reactions. The electronic degrees of freedom are described at the quantum-mechanical level in the Born-Oppenheimer approximation, while the nuclear degrees of freedom are coupled to a thermal bath, through a Langevin equation. This method is based on the path integral representation for the stochastic dynamics and yields the time evolution of both nuclear and electronic degrees of freedom, along the most probable reaction pathways, without spending computational time to explore metastable states. This approach is very efficient and allows to study thermally activated reactions which cannot be simulated using ab-initio molecular dynamics techniques. As a first illustrative application, we characterize the dominant pathway in the cyclobutene to butadiene reaction.
💡 Research Summary
The paper presents a novel first‑principles framework for simulating rare thermally activated chemical reactions, a class of processes that are notoriously difficult to capture with conventional ab‑initio molecular dynamics (AIMD) due to the long time scales required to cross high free‑energy barriers. The authors combine a quantum‑mechanical description of the electronic degrees of freedom, treated within the Born‑Oppenheimer approximation, with a stochastic Langevin dynamics for the nuclei. The nuclear motion obeys the Langevin equation, which incorporates both deterministic forces derived from the electronic potential energy surface (PES) and stochastic forces representing thermal fluctuations and friction. By recasting the stochastic dynamics in a path‑integral formalism, the authors obtain a statistical weight for each possible nuclear trajectory. The most probable reaction pathway—defined as the trajectory that maximizes this weight—is then identified through a variational principle, effectively providing a “most‑likely” reaction coordinate without the need to pre‑define a collective variable or to sample metastable basins exhaustively.
Key technical elements include: (1) a rigorous derivation of the path‑integral representation for Langevin dynamics, leading to an action functional that depends explicitly on the PES, friction coefficient, temperature, and nuclear mass; (2) a practical algorithm that alternates between electronic structure calculations (e.g., density‑functional theory) to update the PES and a deterministic optimization of the action to converge on the optimal trajectory; (3) a treatment of the fluctuation‑dissipation theorem that guarantees the correct equilibrium distribution and allows the user to tune the friction to mimic different solvent or environmental conditions. Because the method focuses solely on the dominant pathway, it avoids the exponential cost associated with brute‑force sampling of rare events, making it orders of magnitude more efficient than standard AIMD or enhanced‑sampling techniques such as metadynamics or umbrella sampling.
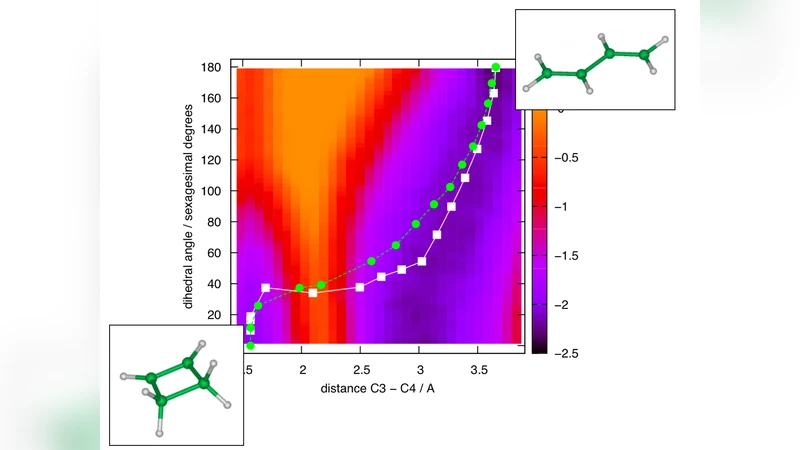
The authors demonstrate the approach on the cyclobutene‑to‑butadiene ring‑opening reaction, a prototypical pericyclic transformation with an experimentally measured activation free energy of roughly 30 kcal mol⁻¹. Using their framework, they recover the correct transition‑state geometry, the associated free‑energy barrier (≈ 31 kcal mol⁻¹), and the detailed electronic reorganization along the pathway—all within a computational effort equivalent to a few hundred femtoseconds of dynamics, far shorter than the nanosecond‑to‑microsecond timescales required by conventional AIMD to observe a single crossing event. The analysis also reveals how the π‑bond formation and σ‑bond cleavage are synchronized with specific changes in the electron density, providing mechanistic insight that aligns with both experimental observations and high‑level quantum‑chemical calculations.
Beyond this benchmark, the paper discusses the broader applicability of the method. By adjusting the temperature and friction parameters, one can model reactions under diverse thermodynamic conditions, from low‑temperature cryogenic environments to high‑temperature combustion processes. The electronic‑structure module is modular, allowing substitution of more accurate post‑DFT methods (e.g., hybrid functionals, GW, or coupled‑cluster) when higher fidelity is required. Potential extensions include surface reactions on catalytic materials, electron‑transfer processes in biological systems, and high‑energy phenomena such as shock‑induced chemistry, all of which suffer from the same sampling bottleneck in traditional AIMD.
In summary, this work introduces a powerful, theoretically sound, and computationally efficient strategy for probing rare, thermally activated reactions at the quantum‑mechanical level. By marrying Langevin stochastic dynamics with a path‑integral variational search for the most probable trajectory, the authors bypass the need for exhaustive sampling of metastable states while retaining full electronic detail. The successful application to the cyclobutene‑butadiene conversion validates the method’s accuracy and efficiency, and the discussion of future directions points to a wide range of scientific problems that could benefit from this approach.
Comments & Academic Discussion
Loading comments...
Leave a Comment