Molecular Systems with Infinite and Finite Degrees of Freedom. Part I: Multi-Scale Analysis
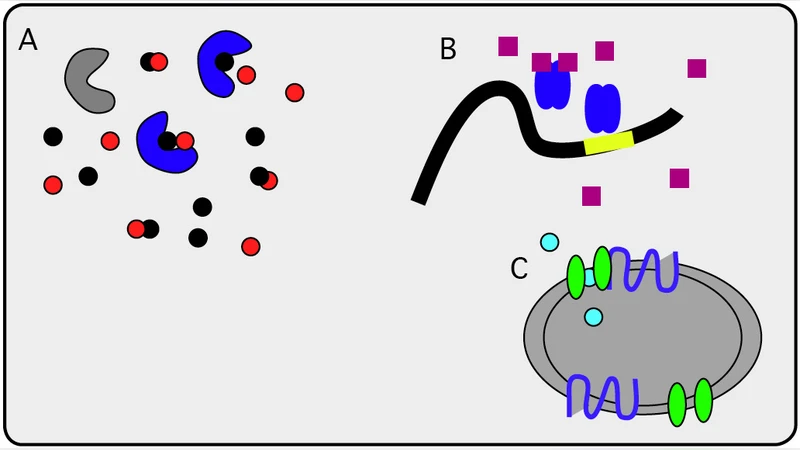
The paper analyses stochastic systems describing reacting molecular systems with a combination of two types of state spaces, a finite-dimensional, and an infinite dimenional part. As a typical situation consider the interaction of larger macro-molecules, finite and small in numbers per cell (like protein complexes), with smaller, very abundant molecules, for example metabolites. We study the construction of the continuum approximation of the associated Master Equation (ME) by using the Trotter approximation [27]. The continuum limit shows regimes where the finite degrees of freedom evolve faster than the infinite ones. Then we develop a rigourous asymptotic adiabatic theory upon the condition that the jump process arising from the finite degrees of freedom of the Markov Chain (MC, typically describing conformational changes of the macro-molecules) occurs with large frequency. In a second part of this work, the theory is applied to derive typical enzyme kinetics in an alternative way and interpretation within this framework.
💡 Research Summary
The paper addresses stochastic reaction networks that involve two fundamentally different types of variables: a large, essentially continuous pool of small molecules (metabolites, ions, etc.) whose copy numbers are high, and a small, discrete set of macro‑molecules (protein complexes, enzymes, receptors) that exist in a few conformational states. The authors formulate a Master Equation (ME) on the product state space ℕⁿ × Σ, where ℕⁿ records the integer counts of the abundant species and Σ is a finite set describing the conformational state of each macro‑molecule. Transition rates are written as products of kinetic constants and functions of the continuous variables, and a small parameter ε is introduced to scale the rates associated with the finite‑state Markov chain (the “fast” conformational jumps).
To pass from the discrete description to a continuum approximation, the authors employ the Trotter product formula, which separates the generator of the full process into two commuting parts: (i) an infinite‑dimensional operator L₀(x,θ) that, after the limit N→∞, becomes a drift‑diffusion (Fokker‑Planck) operator acting on the continuous concentrations x, and (ii) a finite‑dimensional generator Q(θ) multiplied by ε⁻¹ that governs the rapid jumps among the states θ∈Σ. In the limit ε→0 the fast Markov chain equilibrates almost instantly, yielding a conditional stationary distribution π(θ|x).
The central technical contribution is a rigorous adiabatic (or singular perturbation) analysis of the combined system. By treating ε⁻¹ as a large parameter, the authors prove that the joint probability density p(x,θ,t) can be approximated, up to O(ε) errors, by a product of the fast‑equilibrium distribution and a slowly evolving marginal ρ(x,t):
p(x,θ,t) ≈ π(θ|x) ρ(x,t).
The marginal ρ satisfies an effective Fokker‑Planck equation in which the drift and diffusion coefficients are averaged with respect to π. This averaging procedure is justified using semigroup theory, spectral properties of Q, and energy estimates that guarantee strong convergence of the full semigroup to the reduced one.
A key insight is that when the conformational dynamics of the macro‑molecules are much faster than the diffusion of the small species, the macro‑states act as a “hidden” fast variable that modulates the reaction rates of the slow variables. Consequently, the effective kinetic law for the slow species is no longer a simple mass‑action expression; instead, it contains additional nonlinear terms that reflect the probability‑weighted contribution of each conformational state. In particular, the authors show that the classic Michaelis–Menten equation emerges only under the restrictive assumption that the enzyme has a single dominant conformational state. When multiple states are accessible, the adiabatic reduction yields a generalized rate law with extra correction terms that can capture phenomena such as allosteric regulation, substrate inhibition, or cooperative binding.
The paper also discusses the mathematical conditions required for the reduction to hold: the fast generator Q must be irreducible (ensuring a unique stationary distribution), its eigenvalues must be separated from zero by a spectral gap, and the scaling exponents of the reaction rates must satisfy certain balance relations (the authors denote them by α_i). Under these hypotheses, the authors derive explicit error bounds and demonstrate that the reduced model faithfully reproduces the statistics of the full stochastic system in the limit of large molecule numbers and fast conformational switching.
In the companion Part II, the authors apply the framework to concrete enzymatic systems, re‑deriving the Michaelis–Menten kinetics from first principles and showing how the adiabatic correction terms explain deviations observed experimentally in multi‑state enzymes. The methodology provides a systematic way to incorporate discrete conformational dynamics into continuum reaction‑diffusion models, opening new avenues for multiscale modeling of cellular biochemistry where both stochasticity and structural heterogeneity are essential.
Overall, the work delivers a mathematically rigorous multiscale reduction technique for hybrid stochastic systems, clarifies the interplay between infinite and finite degrees of freedom, and offers a fresh perspective on enzyme kinetics that bridges discrete Markov‑chain descriptions with classical continuum approaches.
Comments & Academic Discussion
Loading comments...
Leave a Comment