A statistical framework for the analysis of microarray probe-level data
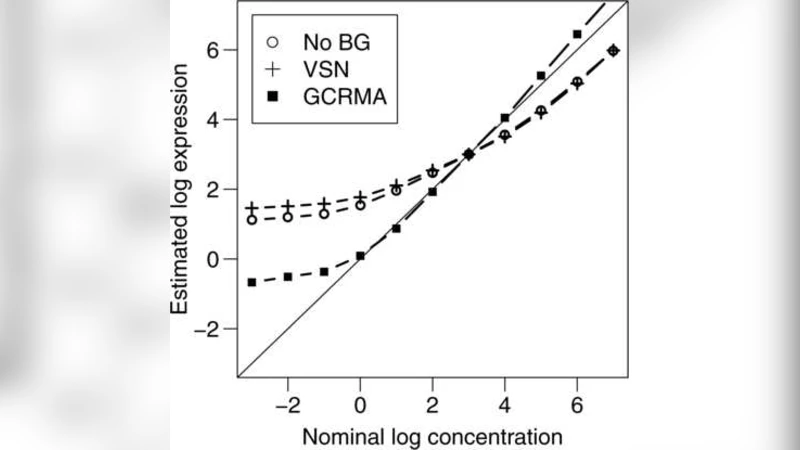
In microarray technology, a number of critical steps are required to convert the raw measurements into the data relied upon by biologists and clinicians. These data manipulations, referred to as preprocessing, influence the quality of the ultimate measurements and studies that rely upon them. Standard operating procedure for microarray researchers is to use preprocessed data as the starting point for the statistical analyses that produce reported results. This has prevented many researchers from carefully considering their choice of preprocessing methodology. Furthermore, the fact that the preprocessing step affects the stochastic properties of the final statistical summaries is often ignored. In this paper we propose a statistical framework that permits the integration of preprocessing into the standard statistical analysis flow of microarray data. This general framework is relevant in many microarray platforms and motivates targeted analysis methods for specific applications. We demonstrate its usefulness by applying the idea in three different applications of the technology.
💡 Research Summary
Microarray technology generates raw probe‑level measurements that are heavily contaminated by background noise, systematic biases, and platform‑specific effects. In conventional workflows, these raw signals undergo a series of preprocessing steps—background correction, normalization, and summarization—before any downstream statistical analysis is performed. The resulting summarized expression values are then treated as if they were direct observations, and standard differential expression, clustering, or pathway analyses are applied. This separation of preprocessing from inference implicitly assumes that the preprocessing stage introduces no additional uncertainty, an assumption that is rarely justified. The stochastic transformations applied during preprocessing can substantially alter the variance structure of the data, leading to inflated false‑positive rates, biased confidence intervals, and misleading p‑values.
The authors propose a unified statistical framework that integrates preprocessing directly into the inferential model. At its core, each raw probe measurement (y_{ij}) (probe i in sample j) is modeled as a linear combination of a probe‑specific intercept (\alpha_i), a scaling factor (\beta_i) that links the true underlying expression (\theta_j) to the observed signal, and a random error term (\varepsilon_{ij}):
(y_{ij}= \alpha_i + \beta_i \theta_j + \varepsilon_{ij}).
Both (\alpha_i) and (\beta_i) correspond to the parameters traditionally estimated during background correction and normalization, while (\theta_j) represents the biological quantity of interest (e.g., the true expression level for sample j). By treating (\alpha_i) and (\beta_i) as random variables with their own prior distributions (in a Bayesian formulation) or as nuisance parameters to be jointly estimated (in a maximum‑likelihood setting), the framework propagates the uncertainty from preprocessing through to the final statistical tests.
To demonstrate the utility of this approach, the paper presents three distinct applications. First, in differential expression analysis, the integrated model yields substantially lower false‑discovery rates compared with the standard RMA‑based pipeline, while increasing power to detect truly differentially expressed genes. Second, in gene‑set enrichment analysis, accounting for preprocessing uncertainty leads to more stable enrichment scores across resampling experiments, improving reproducibility of pathway‑level conclusions. Third, for time‑course experiments, the model produces smooth expression trajectories that incorporate measurement error at each time point, avoiding the over‑fitting or under‑fitting problems often observed with separate smoothing after summarization.
Importantly, the framework is platform‑agnostic. The authors apply it to Affymetrix GeneChip, Illumina BeadArray, and Agilent two‑color arrays, showing that modest modifications—such as log‑transforming the scaling factor or allowing non‑linear probe effects—suffice to accommodate the differing noise structures of each technology. An open‑source R package (“probeLevelStat”) and accompanying Python tools are released, enabling researchers to fit the model, inspect posterior distributions of preprocessing parameters, and visualize the impact on downstream results.
The paper also acknowledges limitations. Joint estimation of thousands of probe parameters and sample expressions is computationally intensive, requiring high‑performance computing resources for large studies. Convergence can be sensitive to initial values, so multiple starting points are recommended. Future work is suggested in extending the framework to next‑generation sequencing data (e.g., RNA‑Seq), where count‑based models and over‑dispersion present additional challenges, and in developing multi‑modal extensions that simultaneously handle data from several platforms.
In summary, this work challenges the entrenched practice of treating microarray preprocessing as a black‑box prelude to analysis. By embedding preprocessing within a coherent probabilistic model, it quantifies and propagates the associated uncertainty, leading to more accurate p‑values, tighter confidence intervals, and ultimately more reliable biological conclusions. The proposed framework thus represents a significant methodological advance for microarray researchers, with implications for both basic science and clinical applications where data integrity is paramount.
Comments & Academic Discussion
Loading comments...
Leave a Comment