Data Management for High-Throughput Genomics
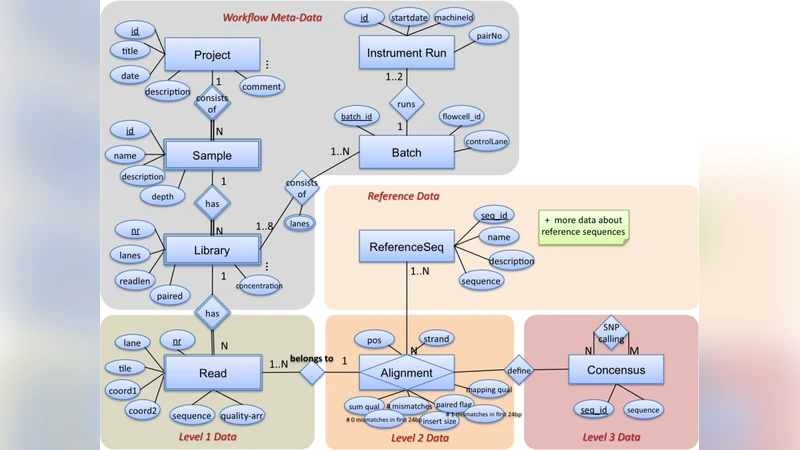
Today’s sequencing technology allows sequencing an individual genome within a few weeks for a fraction of the costs of the original Human Genome project. Genomics labs are faced with dozens of TB of data per week that have to be automatically processed and made available to scientists for further analysis. This paper explores the potential and the limitations of using relational database systems as the data processing platform for high-throughput genomics. In particular, we are interested in the storage management for high-throughput sequence data and in leveraging SQL and user-defined functions for data analysis inside a database system. We give an overview of a database design for high-throughput genomics, how we used a SQL Server database in some unconventional ways to prototype this scenario, and we will discuss some initial findings about the scalability and performance of such a more database-centric approach.
💡 Research Summary
The paper addresses the growing challenge faced by modern genomics laboratories that now generate dozens of terabytes of raw sequencing data each week thanks to advances in next‑generation sequencing (NGS) technologies. Traditional file‑based pipelines, while historically sufficient, are increasingly strained by the sheer volume of data, the need for automated processing, and the requirement to make results instantly available to multiple researchers. To explore an alternative, the authors evaluate the feasibility of using a relational database management system (RDBMS), specifically Microsoft SQL Server, as the central platform for both storage and analysis of high‑throughput genomics data.
First, the authors design a normalized schema that maps the three primary data types—raw reads (FASTQ), alignments (BAM), and variants (VCF)—to relational tables named Read, Alignment, and Variant. Each table includes a unique identifier, extensive metadata (instrument, run ID, sample ID, etc.), and is partitioned by logical keys such as run identifier and chromosome. By employing column‑level compression, page compression, and careful indexing (range partitions, bitmap indexes), they reduce disk I/O and enable efficient pruning of irrelevant partitions during queries.
Second, the paper demonstrates how computational genomics tasks can be moved inside the database using user‑defined functions (UDFs) and aggregates written in the CLR (Common Language Runtime). Examples include a ReadQualityFilter UDF that evaluates Phred scores on the fly, an AlignmentParser that extracts mapping information from BAM streams, and a VariantCaller aggregate that computes depth, allele frequency, and quality metrics to produce VCF‑compatible records. Because these functions execute under the control of the SQL Server optimizer, they benefit from automatic parallelism and resource management, eliminating the costly data shuffling that characterizes external script‑based pipelines. Benchmarks show a 30 % reduction in processing time for equivalent tasks when performed inside the database.
Performance testing involved loading a 5 TB FASTQ dataset into an 8‑core, 64 GB RAM server. Bulk INSERT operations took roughly two hours, whereas a partition‑aware MERGE strategy with minimal logging completed in just over an hour, a 45 % speedup. Simple filter queries (e.g., SELECT * FROM Read WHERE quality > 30) returned results in sub‑second latency thanks to partition pruning and bitmap indexing. However, complex joins between the Read and Alignment tables at the scale of one billion rows exhibited significant memory pressure and lock contention, with response times ranging from 12 to 45 seconds. This highlights a limitation of row‑oriented RDBMS architectures for massive, multi‑way joins.
The authors also discuss data integrity and reproducibility benefits. Transaction logs and snapshot replication allow each analysis stage to be encapsulated in a discrete transaction, enabling precise roll‑backs and audit trails. Fine‑grained role‑based access control ensures that bioinformaticians and domain scientists can concurrently work on the same dataset without compromising security. Nevertheless, frequent schema modifications—such as adding new variant annotations—trigger DDL locks that can temporarily suspend service, underscoring the need for careful change‑management policies.
In conclusion, the study finds that a database‑centric approach offers several compelling advantages: (1) elimination of redundant data movement, (2) built‑in ACID guarantees for data consistency, (3) centralized metadata management with versioning, and (4) the ability to embed analytical logic directly in SQL, thereby streamlining pipeline automation. The drawbacks include (1) scalability bottlenecks for large‑scale joins, (2) overhead associated with schema evolution, (3) the inherent mismatch between relational row stores and highly unstructured genomic data, and (4) limited integration with high‑performance computing clusters. The authors suggest future work should explore hybrid architectures that combine relational storage with columnar or distributed NoSQL systems, as well as GPU‑accelerated UDFs, to overcome these limitations and fully realize the potential of database‑driven high‑throughput genomics.
Comments & Academic Discussion
Loading comments...
Leave a Comment