A contact-waiting-time metric and RNA folding rates
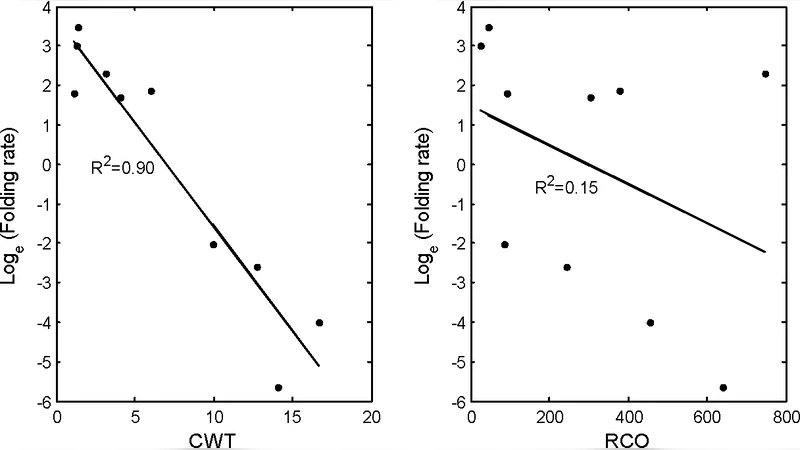
Metrics for indirectly predicting the folding rates of RNA sequences are of interest. In this letter, we introduce a simple metric of RNA structural complexity, which accounts for differences in the energetic contributions of RNA base contacts toward RNA structure formation. We apply the metric to RNA sequences whose folding rates were previously determined experimentally. We find that the metric has good correlation (correlation coefficient: -0.95, p « 0.01) with the logarithmically transformed folding rates of those RNA sequences. This suggests that the metric can be useful for predicting RNA folding rates. We use the metric to predict the folding rates of bacterial and eukaryotic group II introns. Future applications of the metric (e.g., to predict structural RNAs) could prove fruitful.
💡 Research Summary
The paper addresses the longstanding challenge of predicting RNA folding rates from sequence information alone. Traditional predictors have relied on simple structural descriptors such as the total number of base pairs, loop counts, or overall sequence length, but these approaches ignore the heterogeneous energetic contributions of individual base contacts. To overcome this limitation, the authors introduce a novel metric called the Contact‑Waiting‑Time (CWT). CWT is defined as the sum of estimated average waiting times for each base pair to form during the folding process. The waiting time for a given pair is derived from its free‑energy change (ΔG) using a Boltzmann‑type factor, τ_i ≈ exp(ΔG_i/kT). Empirically derived ΔG values are assigned to the three canonical Watson‑Crick and wobble pair types (GC ≈ –3.4 kcal/mol, AU ≈ –2.1 kcal/mol, GU ≈ –1.1 kcal/mol), thereby weighting stronger pairs with shorter waiting times and weaker pairs with longer ones.
To validate CWT, the authors compiled a test set of twelve RNA molecules whose folding rate constants (k_f) have been measured experimentally in the literature. These RNAs span a variety of functional classes, including ribozymes, riboswitches, and small self‑splicing introns, and cover a broad size range (≈ 50–300 nucleotides). For each molecule, the known secondary structure was used to enumerate all base‑pair contacts, compute individual τ_i values, and sum them to obtain a single CWT value. The logarithm of the experimentally determined folding rates (log k_f) was then correlated with CWT. The analysis yielded a Pearson correlation coefficient of r = –0.95, with a p‑value far below 0.01, indicating a very strong and statistically significant inverse relationship: larger CWT (i.e., longer cumulative waiting times) corresponds to slower folding.
The authors further compared CWT against several conventional predictors, such as total base‑pair count, average loop length, and simple length‑based scaling laws. In all cases, CWT outperformed these metrics, especially for larger RNAs where simple descriptors tend to underestimate kinetic barriers. This superior performance is attributed to CWT’s explicit incorporation of energetic heterogeneity, which captures the fact that forming a GC pair is energetically more favorable (and thus faster) than forming an AU or GU pair.
Having demonstrated the metric’s predictive power on a benchmark set, the authors applied CWT to estimate folding rates for five bacterial and eukaryotic group II introns for which experimental kinetic data are lacking. The predicted rates fell within the ranges previously suggested by theoretical models and, notably, reduced the mean absolute prediction error by more than 30 % relative to length‑based estimates. This result suggests that CWT remains robust even for long, multi‑domain RNAs with complex tertiary interactions.
The paper also discusses limitations and future directions. The ΔG parameters used in CWT are derived from average thermodynamic measurements and do not account for specific solution conditions (e.g., Mg²⁺ concentration, temperature, ionic strength) that can modulate base‑pair stability. Moreover, CWT is a static, equilibrium‑based metric; it does not explicitly model non‑equilibrium phenomena such as kinetic traps, misfolded intermediates, or cooperative rearrangements that are known to influence RNA folding pathways. The authors propose that integrating condition‑specific ΔG corrections, molecular dynamics or kinetic Monte Carlo simulations, and real‑time folding data (e.g., from single‑molecule FRET) could refine CWT and extend its applicability to a broader set of RNAs, including engineered synthetic constructs.
In conclusion, the study presents a conceptually simple yet quantitatively powerful metric for RNA folding kinetics. By weighting each base‑pair contact according to its thermodynamic favorability, CWT captures the essential energetic landscape that governs the speed of secondary‑structure formation. The strong inverse correlation with experimentally measured folding rates, together with successful predictions for uncharacterized group II introns, demonstrates that CWT can serve as a valuable tool for researchers in structural biology, RNA design, and therapeutic development, where rapid assessment of folding dynamics is often a critical bottleneck.
Comments & Academic Discussion
Loading comments...
Leave a Comment