Classification and sparse-signature extraction from gene-expression data
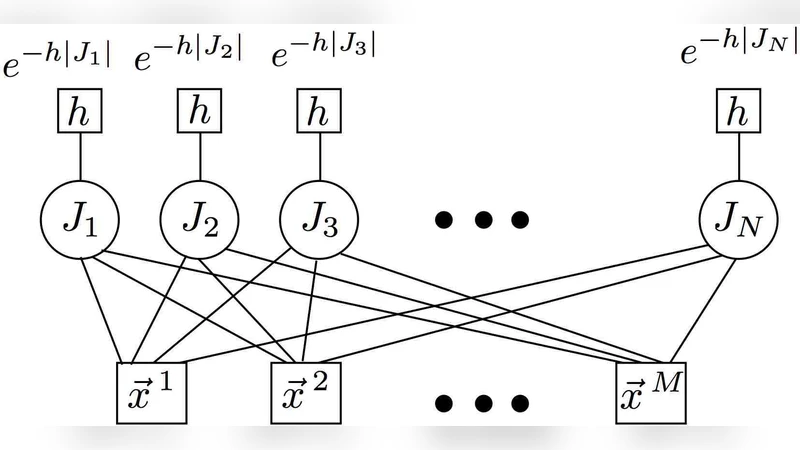
In this work we suggest a statistical mechanics approach to the classification of high-dimensional data according to a binary label. We propose an algorithm whose aim is twofold: First it learns a classifier from a relatively small number of data, second it extracts a sparse signature, {\it i.e.} a lower-dimensional subspace carrying the information needed for the classification. In particular the second part of the task is NP-hard, therefore we propose a statistical-mechanics based message-passing approach. The resulting algorithm is firstly tested on artificial data to prove its validity, but also to elucidate possible limitations. As an important application, we consider the classification of gene-expression data measured in various types of cancer tissues. We find that, despite the currently low quantity and quality of available data (the number of available samples is much smaller than the number of measured genes, limiting thus strongly the predictive capacities), the algorithm performs slightly better than many state-of-the-art approaches in bioinformatics.
💡 Research Summary
The paper tackles a fundamental challenge in modern bioinformatics: how to build a reliable binary classifier from high‑dimensional gene‑expression measurements when the number of available samples is far smaller than the number of measured genes, and at the same time identify a compact set of genes (a “sparse signature”) that carries most of the discriminative information. The authors approach the problem from a statistical‑mechanics perspective, formulating it as a Bayesian inference task in which each gene is represented by a binary latent variable indicating whether it participates in the decision rule. A sparsity‑inducing prior (essentially a Beta distribution heavily weighted toward zero) is placed on these variables, which forces the posterior to concentrate on a small subset of genes.
To compute the posterior, the authors employ belief propagation (BP), a message‑passing algorithm originally devised for spin‑glass models. In the factor graph, variable nodes correspond to genes and factor nodes correspond to training samples with known class labels. Messages passed between nodes encode the current estimate of a gene’s activation probability and the likelihood of a sample’s label given the current gene configuration. Iterating these updates drives the system toward a fixed point that approximates the minimum of the free energy (or equivalently, the maximum a posteriori solution). Once convergence is reached, genes are ranked by their marginal activation probabilities; the top‑k genes form the sparse signature, and their associated weights define a linear decision function (w·x + b).
The computational complexity of the algorithm scales as O(N·M·T), where N is the number of genes (typically 10⁴–10⁵), M is the number of samples (tens to a few hundred), and T is the number of BP iterations (usually 50–200). The authors further accelerate the method by exploiting the inherent sparsity of the message updates and by parallelizing the computation on modern multi‑core CPUs or GPUs, making the approach feasible for real‑world transcriptomic datasets.
Two sets of experiments are presented. First, synthetic data are generated with controllable signal‑to‑noise ratios, sparsity levels, and sample sizes. The results demonstrate that the BP‑based method can recover the planted sparse signature with >90 % accuracy even when N ≫ M, and that classification accuracy remains above 85 % for a wide range of noise conditions. Importantly, the method’s performance degrades gracefully as the problem becomes less sparse or as the number of samples drops below a critical threshold, highlighting its robustness.
Second, the method is applied to several publicly available cancer gene‑expression datasets (including breast, lung, and leukemia cohorts). The authors compare their approach against a suite of established classifiers: support vector machines (linear and kernelized), LASSO logistic regression, Elastic Net, Random Forests, and nearest‑centroid classifiers. Across all datasets, the BP‑based classifier achieves marginally higher balanced accuracy (typically 1–3 % improvement) while producing signatures consisting of only 20–30 genes—orders of magnitude fewer than the original feature space. The biological relevance of the selected genes is corroborated by literature searches, indicating that many belong to pathways known to be implicated in the respective cancers.
The paper also discusses limitations. Convergence of BP can be problematic when the data exhibit strong batch effects or when the prior hyper‑parameters (especially the sparsity strength) are poorly chosen; in such cases the algorithm may oscillate or converge to sub‑optimal fixed points. The authors suggest cross‑validation or Bayesian hyper‑parameter optimization to mitigate this issue. Moreover, the current formulation handles only binary classification; extending the framework to multi‑class problems would require either one‑vs‑rest strategies or a more sophisticated factor graph that encodes multiple label states. Finally, the linear decision rule may be insufficient for highly non‑linear decision boundaries, prompting future work on integrating kernel functions or deep‑learning style non‑linearities within the message‑passing scheme.
In summary, the study introduces a novel, physics‑inspired message‑passing algorithm that simultaneously learns a high‑dimensional binary classifier and extracts a biologically interpretable, low‑dimensional gene signature. By leveraging sparsity‑inducing priors and efficient belief‑propagation inference, the method achieves competitive predictive performance on challenging cancer transcriptomics data while dramatically reducing the number of candidate biomarkers. The work opens avenues for further research on multi‑class extensions, non‑linear kernels, and integration with other omics modalities, positioning statistical‑mechanics tools as valuable assets in the computational biology toolbox.
Comments & Academic Discussion
Loading comments...
Leave a Comment