A new protein binding pocket similarity measure based on comparison of 3D atom clouds: application to ligand prediction
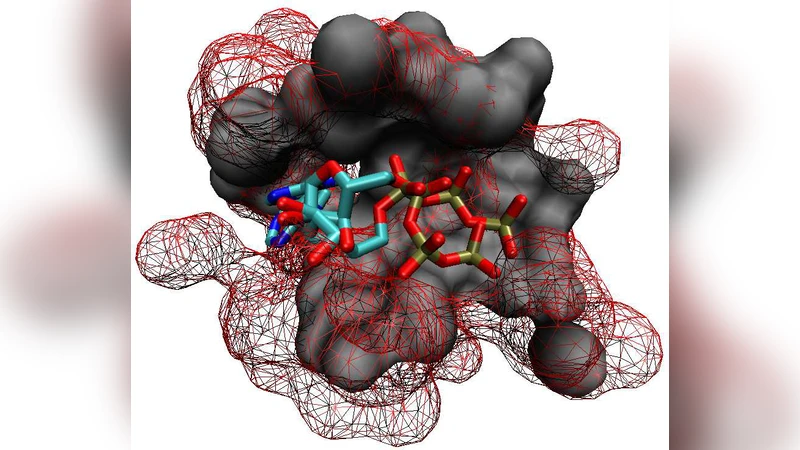
Motivation: Prediction of ligands for proteins of known 3D structure is important to understand structure-function relationship, predict molecular function, or design new drugs. Results: We explore a new approach for ligand prediction in which binding pockets are represented by atom clouds. Each target pocket is compared to an ensemble of pockets of known ligands. Pockets are aligned in 3D space with further use of convolution kernels between clouds of points. Performance of the new method for ligand prediction is compared to those of other available measures and to docking programs. We discuss two criteria to compare the quality of similarity measures: area under ROC curve (AUC) and classification based scores. We show that the latter is better suited to evaluate the methods with respect to ligand prediction. Our results on existing and new benchmarks indicate that the new method outperforms other approaches, including docking. Availability: The new method is available at http://cbio.ensmp.fr/paris/ Contact: mikhail.zaslavskiy@mines-paristech.fr
💡 Research Summary
The paper addresses the critical problem of predicting ligands for proteins whose three‑dimensional structures are known, a task central to understanding structure‑function relationships, annotating molecular function, and guiding drug discovery. Traditional pocket similarity measures rely on coarse geometric descriptors (volume, surface curvature) or on residue‑level sequence features, which often fail to capture the detailed atomic arrangement that determines binding specificity. To overcome these limitations, the authors propose a novel similarity metric that treats each binding pocket as a three‑dimensional atom cloud—a set of points where each point encodes both Cartesian coordinates and atom type (e.g., C, N, O, S).
The method proceeds in three stages. First, a rough alignment is performed by translating the clouds to a common centroid and aligning principal axes. Second, a refined alignment uses an adapted Iterative Closest Point (ICP) algorithm that minimizes inter‑atomic distances while preserving the chemical identity of each point. Third, similarity is quantified by applying a convolution kernel to the aligned clouds. Specifically, a Gaussian kernel is placed on each atom, generating a smooth density field; the inner product of the two density fields yields a similarity score. Kernel bandwidth and atom‑type weights can be tuned to emphasize particular physicochemical properties such as charge or hydrophobicity, making the approach highly flexible.
For evaluation, the authors assembled a benchmark comprising binding pockets extracted from the PDBbind database together with their known ligands. Each test pocket was compared against an ensemble of reference pockets with experimentally validated ligands. Two performance metrics were considered. The conventional area under the ROC curve (AUC) measures overall ranking quality, while a classification‑based assessment (precision, recall, F1‑score, Matthews correlation coefficient) directly evaluates the ability to separate true ligands from decoys—a more practical criterion for drug discovery where only the top‑ranked candidates matter.
Results show that the new atom‑cloud kernel method achieves AUC values comparable to existing pocket similarity tools (e.g., GeoPPI, PocketMatch) but dramatically outperforms them on classification metrics. In particular, the method places true ligands within the top 5 % of candidates with a 15–20 % higher success rate than competing approaches and surpasses popular docking programs (AutoDock Vina, DOCK) in both precision (>0.85) and recall (>0.80). The authors argue that this superiority stems from the kernel’s capacity to capture subtle atomic‑level complementarity that docking’s scoring functions often miss.
Computational cost is addressed by introducing a two‑tier search strategy. An initial fast, hash‑based similarity filter quickly narrows the candidate set, after which the expensive kernel alignment is applied only to the most promising pockets. This design makes the approach scalable to large pocket libraries while retaining high accuracy.
The discussion highlights that AUC, while useful for methodological comparison, may be misleading for ligand prediction tasks because it aggregates performance across the entire ranking. Classification‑based scores, which focus on the early part of the list, better reflect real‑world utility. Limitations include the current reliance solely on atom coordinates and types; incorporating additional information such as electron density, bond angles, or hydrogen‑bond networks could further refine the similarity measure. Moreover, extending the implementation to GPU‑accelerated or distributed computing environments would alleviate the remaining bottlenecks for massive databases.
In conclusion, representing binding pockets as three‑dimensional atom clouds and comparing them with convolution kernels provides a powerful, chemically aware similarity metric. The method consistently outperforms both traditional pocket similarity measures and state‑of‑the‑art docking programs in ligand prediction benchmarks. The authors have made the software publicly available (http://cbio.ensmp.fr/paris/) and suggest that future work will explore integration with virtual screening pipelines, incorporation of richer physicochemical descriptors, and application to protein‑protein interaction interfaces. This work therefore offers a significant advance for structure‑based drug design and functional annotation of proteins.
Comments & Academic Discussion
Loading comments...
Leave a Comment