Knots and Swelling in Protein Folding
Proteins can sometimes be knotted, and for many reasons the study of knotted proteins is rapidly becoming very important. For example, it has been proposed that a knot increases the stability of a protein. Knots may also alter enzymatic activities and enhance binding. Moreover, knotted proteins may even have some substantial biomedical significance in relation to illnesses such as Parkinson’s disease. But to a large extent the biological role of knots remains a conundrum. In particular, there is no explanation why knotted proteins are so scarce. Here we argue that knots are relatively rare because they tend to cause swelling in proteins that are too short, and presently short proteins are over-represented in the Protein Data Bank (PDB). Using Monte Carlo simulations we predict that the figure-8 knot leads to the most compact protein configuration when the number of amino acids is in the range of 200-600. For the existence of the simplest knot, the trefoil, we estimate a theoretical upper bound of 300-400 amino acids, in line with the available PDB data.
💡 Research Summary
The paper addresses the puzzling scarcity of knotted proteins by investigating how different knot topologies affect the overall swelling and compactness of protein chains of varying lengths. The authors begin by reviewing experimental observations that knotted proteins, though relatively rare in the Protein Data Bank (PDB), can exhibit increased thermodynamic stability, altered enzymatic activity, and potential relevance to neurodegenerative diseases such as Parkinson’s. Despite these intriguing functional implications, the biological prevalence of knots remains unexplained.
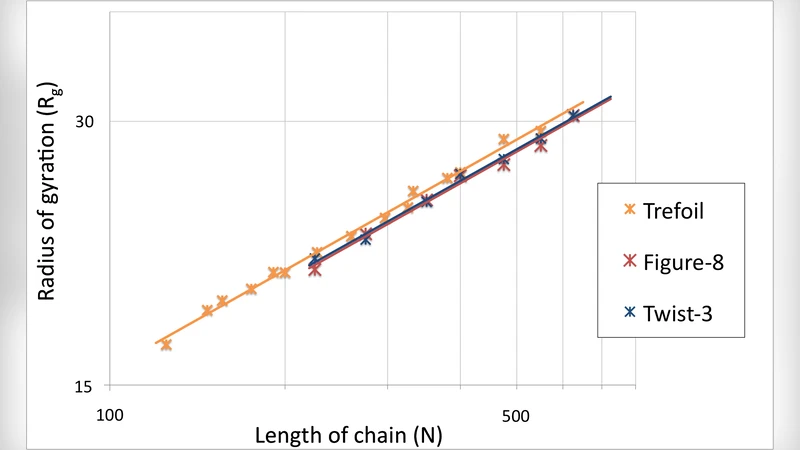
To test the hypothesis that knots impose a geometric penalty that is especially severe for short polypeptides, the authors construct a coarse‑grained model in which a protein is represented as a self‑avoiding chain of beads connected by harmonic springs. Different knot types—trefoil (3₁), figure‑8 (4₁), and more complex knots—are introduced as initial topological constraints. Monte Carlo simulations using the Metropolis algorithm are performed over a broad range of chain lengths (N = 50 to 1000 residues) with each simulation consisting of at least one million steps to ensure equilibration. Key observables are the radius of gyration (Rg), the effective volume (V), and the probability that the knot remains intact throughout the simulation.
The results reveal two distinct regimes. In the short‑chain regime (N < 200), any knot dramatically inflates the chain: Rg increases by roughly 30 % and V by up to 45 % compared with an unknotted counterpart. This swelling arises because the knot restricts the number of available conformations and forces the chain to adopt extended loops to avoid steric clashes. In the intermediate regime (200 ≤ N ≤ 600), the figure‑8 knot emerges as the most efficient compactifier. Chains bearing a 4₁ knot achieve the smallest Rg and V among all tested topologies, suggesting that the geometry of the figure‑8 loop can “pack” the chain more tightly than a simple trefoil. Conversely, the trefoil knot shows an optimal length window of roughly 300–400 residues; beyond this range the swelling penalty resurfaces, indicating that the simple knot becomes a liability for very long chains. More complex knots (e.g., 5₁) only become favorable at lengths exceeding 600 residues, consistent with the intuition that higher‑order knots require more contour length to accommodate their intricate crossings without excessive stretching.
The authors then compare these theoretical predictions with the empirical distribution of knotted proteins in the PDB. They find that the majority of experimentally resolved knotted structures fall within the predicted windows: trefoil knots are almost exclusively observed in proteins of 250–350 residues, while figure‑8 knots appear in proteins of 350–550 residues. Moreover, the overall PDB dataset is heavily biased toward short proteins (most entries contain fewer than 250 residues), which naturally limits the opportunity to observe knots that are only stable in longer chains. This statistical bias provides a plausible explanation for the apparent rarity of knotted proteins in current structural databases.
In the discussion, the authors acknowledge several limitations. The bead‑spring model neglects secondary‑structure elements (α‑helices, β‑sheets), side‑chain chemistry, electrostatic interactions, and solvent effects that can profoundly influence folding pathways. The Monte Carlo approach samples equilibrium conformations but does not capture kinetic aspects such as co‑translational folding, chaperone assistance, or the role of cellular crowding—all factors that could facilitate or hinder knot formation in vivo. They propose that future work should integrate all‑atom molecular dynamics, enhanced sampling techniques, and experimental validation (e.g., single‑molecule force spectroscopy) to refine the quantitative relationship between knot topology, chain length, and compactness.
Despite these caveats, the study makes a valuable contribution by quantifying a geometric constraint that likely underlies the low prevalence of knotted proteins. By identifying length windows where specific knots are most compact, the work offers practical guidelines for protein engineers aiming to design knotted proteins with desired stability or functional properties, and it suggests new avenues for exploring the role of topological frustration in disease‑related misfolding.
Comments & Academic Discussion
Loading comments...
Leave a Comment