Exploring the Free Energy Landscape: From Dynamics to Networks and Back
The knowledge of the Free Energy Landscape topology is the essential key to understand many biochemical processes. The determination of the conformers of a protein and their basins of attraction takes a central role for studying molecular isomerization reactions. In this work, we present a novel framework to unveil the features of a Free Energy Landscape answering questions such as how many meta-stable conformers are, how the hierarchical relationship among them is, or what the structure and kinetics of the transition paths are. Exploring the landscape by molecular dynamics simulations, the microscopic data of the trajectory are encoded into a Conformational Markov Network. The structure of this graph reveals the regions of the conformational space corresponding to the basins of attraction. In addition, handling the Conformational Markov Network, relevant kinetic magnitudes as dwell times or rate constants, and the hierarchical relationship among basins, complete the global picture of the landscape. We show the power of the analysis studying a toy model of a funnel-like potential and computing efficiently the conformers of a short peptide, the dialanine, paving the way to a systematic study of the Free Energy Landscape in large peptides.
💡 Research Summary
The paper introduces a novel computational framework that translates molecular dynamics (MD) trajectories into a Conformational Markov Network (CMN) in order to characterize the topology and kinetics of a free‑energy landscape (FEL). The authors begin by highlighting the central role of FELs in governing biochemical processes such as protein folding, isomerization, and ligand binding, and they point out that traditional approaches—energy minimization, clustering, or dimensionality reduction—often rely on subjective criteria and can obscure kinetic information. To overcome these limitations, the MD trajectory is discretized into a set of microstates (nodes) using a regular grid or clustering algorithm; directed edges are added for each observed transition between successive frames, and the edge weights count the number of such transitions. Normalizing the transition counts yields a stochastic transition matrix that defines a Markov chain on the network.
Meta‑stable conformers (basins) are identified as strongly connected sub‑graphs where intra‑basin transitions dominate over inter‑basin jumps. Community‑detection methods (e.g., Louvain, Infomap) or eigenvector analysis of the transition matrix are employed to extract these basins automatically. The self‑loop weight of a basin—probability of remaining in the same basin during a time step—provides a direct estimate of the average dwell time, which is inversely related to the depth of the corresponding free‑energy well. Inter‑basin transition rates are read directly from the off‑diagonal elements of the transition matrix, and optimal transition pathways are obtained by applying graph‑theoretic algorithms such as minimum‑resistance (inverse rate) paths or maximum‑flow calculations.
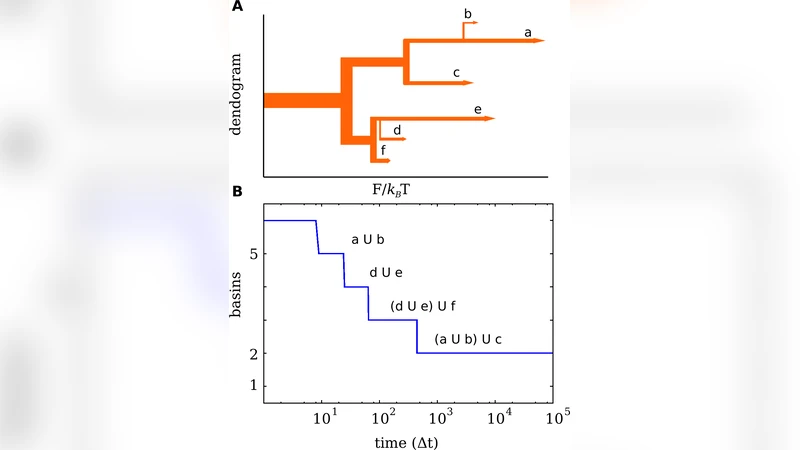
A hierarchical organization of basins emerges naturally when the inter‑basin transition rates are visualized as a dendrogram: basins with high mutual transition rates cluster together at higher levels, while more isolated basins appear as leaf nodes. This hierarchy reflects the multi‑scale nature of FELs, where deep funnels contain nested sub‑funnels.
The methodology is validated on two systems. First, a synthetic funnel‑shaped potential with three predefined minima is used as a toy model. The CMN reconstruction recovers exactly the three basins, their relative depths, and the correct transition barriers, demonstrating that the approach can faithfully reproduce a known landscape. Second, the authors apply the framework to the alanine dipeptide (dialanine) peptide, a classic benchmark for conformational analysis. A 200 ns MD simulation is discretized in the φ‑ψ dihedral space, producing a CMN that automatically identifies the three well‑known meta‑stable states (α‑helix, β‑turn, and a transition basin). Dwell times and rate constants extracted from the network agree with values reported in the literature, and the most probable transition pathway between α‑helix and β‑turn is recovered by the minimum‑resistance algorithm.
Key contributions of the work include: (1) a unified representation of FEL topology and kinetics through a single graph structure; (2) quantitative metrics for basin stability (dwell time) and inter‑basin kinetics (rate constants); (3) an automatic, parameter‑light method for detecting hierarchical relationships among conformers; and (4) demonstration of scalability from simple potentials to realistic peptide systems.
The authors acknowledge several limitations. The choice of discretization resolution (grid spacing or clustering parameters) can influence the number and shape of identified basins, potentially leading to over‑ or under‑segmentation. Rare transition events require long simulation times to be sampled adequately, which may be computationally demanding. For very large proteins or complexes, the resulting CMN can become massive, challenging memory and processing resources. To address these issues, the paper suggests adaptive discretization schemes, enhanced‑sampling techniques such as metadynamics or accelerated MD, and parallel graph‑processing frameworks.
In conclusion, by mapping MD data onto a Markov network, the study provides a powerful tool for extracting both thermodynamic and kinetic information from free‑energy landscapes without resorting to arbitrary clustering thresholds. The approach is poised to impact a broad range of fields, from protein engineering and drug design to the study of large biomolecular assemblies, where understanding the hierarchy of meta‑stable states and the pathways connecting them is essential.
Comments & Academic Discussion
Loading comments...
Leave a Comment