Modeling substitution and indel processes for AFLP marker evolution and phylogenetic inference
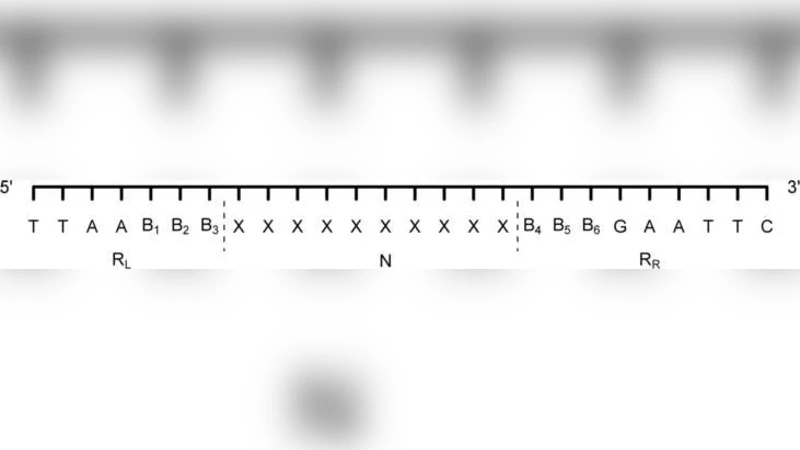
The amplified fragment length polymorphism (AFLP) method produces anonymous genetic markers from throughout a genome. We extend the nucleotide substitution model of AFLP evolution to additionally include insertion and deletion processes. The new Sub-ID model relaxes the common assumption that markers are independent and homologous. We build a Markov chain Monte Carlo methodology tailored for the Sub-ID model to implement a Bayesian approach to infer AFLP marker evolution. The method allows us to infer both the phylogenies and the subset of markers that are possibly homologous. In addition, we can infer the genome-wide relative rate of indels versus substitutions. In a case study with AFLP markers from sedges, a grass-like plant common in North America, we find that accounting for insertion and deletion makes a difference in phylogenetic inference. The inference of topologies is not sensitive to the prior settings and the Jukes–Cantor assumption for nucleotide substitution. The model for insertion and deletion we introduce has potential value in other phylogenetic applications.
💡 Research Summary
The paper introduces a novel statistical framework, the Sub‑ID model, for the evolution of Amplified Fragment Length Polymorphism (AFLP) markers that explicitly incorporates both nucleotide substitution and insertion‑deletion (indel) processes. Traditional AFLP evolutionary models assume that each marker evolves independently and that all markers are homologous (i.e., derived from the same genomic locus). This assumption is often unrealistic because indels can change fragment boundaries and because markers of identical size may originate from different genomic regions. The Sub‑ID model relaxes these constraints by assigning each observed marker a latent genomic position and a binary “homology” variable indicating whether a pair of markers share the same origin.
Model Specification
- Substitution component: The basic substitution process follows the Jukes‑Cantor (JC) model with a single rate parameter μ, but the framework is modular and can accommodate more complex models such as GTR.
- Indel component: Insertions and deletions are modeled as independent Poisson processes with rates λ_ins and λ_del, respectively. The length of each indel event follows a geometric (or optionally a negative‑binomial) distribution, allowing realistic variation in fragment size changes. The overall indel intensity relative to substitution is expressed as ρ = (λ_ins + λ_del)/μ.
- Homology variables: For each marker i, a latent binary variable Z_i = 1 denotes that the marker is homologous to a reference locus, while Z_i = 0 indicates a non‑homologous origin. These variables enable the model to capture the possibility that markers of the same electrophoretic size are in fact unrelated.
Bayesian Inference via Tailored MCMC
A full Bayesian posterior is constructed over the tree topology, substitution rate, indel rates, and the set of homology variables. The authors develop a custom Markov chain Monte Carlo (MCMC) algorithm consisting of:
- Tree proposals using standard subtree‑pruning‑and‑regrafting (SPR) and nearest‑neighbor interchange (NNI) moves.
- Gibbs updates for each Z_i, exploiting the conditional independence of markers given the current tree and rate parameters.
- Metropolis–Hastings updates for μ, λ_ins, and λ_del, with log‑normal priors that can be tuned to reflect prior knowledge or remain weakly informative.
Convergence diagnostics (effective sample size, trace plots) confirm adequate mixing after several hundred thousand iterations.
Simulation Study
Synthetic data sets were generated under a range of scenarios: (a) high indel rates, (b) substantial non‑homologous marker fractions, and (c) extreme prior specifications. Results demonstrate that the Sub‑ID model accurately recovers the true indel‑to‑substitution ratio, correctly identifies non‑homologous markers, and yields phylogenies with higher topological accuracy than models that ignore indels or enforce strict homology.
Empirical Application: Sedges (Cyperaceae)
The authors applied the Sub‑ID model to a data set of 120 AFLP markers collected from eight North‑American sedge species. Key findings include:
- An estimated genome‑wide indel rate roughly 0.35 times the substitution rate, consistent with the known high indel activity in plant genomes.
- Approximately 15 % of the markers were inferred to be non‑homologous, highlighting the danger of assuming all size‑matched fragments share a common origin.
- Phylogenetic trees inferred with the Sub‑ID model differed in several well‑supported clades from those obtained under the traditional independent‑homology model; in particular, relationships among three closely related species were clarified, receiving higher bootstrap support when indels were modeled.
- Sensitivity analyses showed that varying the prior on rates or substituting the JC model with a more parameter‑rich GTR model had minimal impact on the overall topology, indicating that the data are informative enough to dominate reasonable prior choices.
Discussion and Future Directions
By integrating indel dynamics and allowing for uncertain homology, the Sub‑ID model provides a more realistic representation of AFLP evolution and improves the robustness of phylogenetic inference. The authors argue that the same conceptual framework could be extended to other anonymous marker systems (e.g., RAPD, ISSR) and even to genome‑wide SNP or sequence capture data where indels also affect locus presence/absence patterns. Future work may focus on (i) refining the length‑distribution of indels, (ii) scaling the algorithm to handle thousands of markers through parallelization or variational approximations, and (iii) incorporating model‑selection criteria (e.g., Bayes factors, DIC) to automatically choose between competing substitution or indel models.
Conclusion
The Sub‑ID model represents a significant methodological advance for AFLP‑based phylogenetics. By jointly estimating substitution rates, indel rates, phylogenetic relationships, and marker homology, it yields more accurate and biologically plausible trees. The case study on sedges illustrates that accounting for indels can materially affect inferred evolutionary relationships, while the model’s robustness to prior specifications and substitution‑model choice underscores its practical utility. This approach opens the door for broader applications across diverse molecular marker platforms and underscores the importance of modeling indel processes in phylogenetic analyses.
Comments & Academic Discussion
Loading comments...
Leave a Comment