CLeFAPS: Fast Flexible Alignment of Protein Structures Based on Conformational Letters
CLeFAPS, a fast and flexible pairwise structural alignment algorithm based on a rigid-body framework, namely CLePAPS, is proposed. Instead of allowing twists (or bends), the flexible in CLeFAPS means: (a) flexibilization of the algorithm’s parameters through self-adapting with the input structures’ size, (b) flexibilization of adding the aligned fragment pairs (AFPs) into an one-to-multi correspondence set instead of checking their position conflict, (c) flexible fragment may be found through an elongation procedure rooted in a vector-based score instead of a distance-based score. We perform a comparison between CLeFAPS and other popular algorithms including rigid-body and flexible on a closely-related protein benchmark (HOMSTRAD) and a distantly-related protein benchmark (SABmark) while the latter is also for the discrimination test, the result shows that CLeFAPS is competitive with or even outperforms other algorithms while the running time is only 1/150 to 1/50 of them.
💡 Research Summary
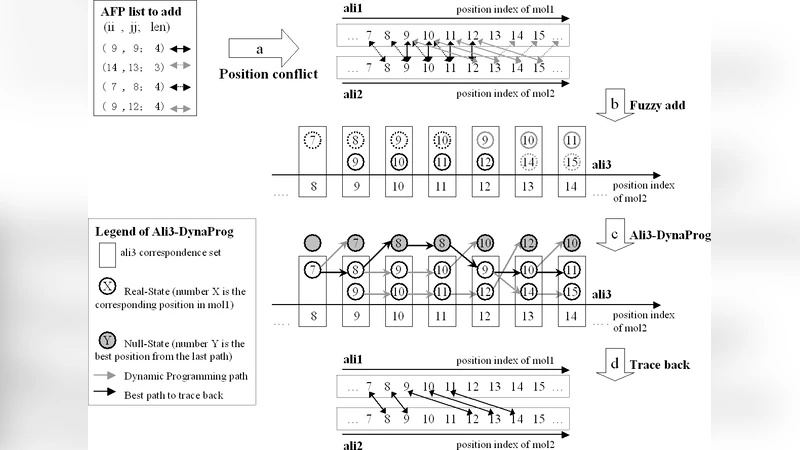
CLeFAPS (Conformational Letter based Fast Flexible Alignment of Protein Structures) is a novel pair‑wise structural alignment algorithm that builds upon the rigid‑body framework CLePAPS while introducing three distinct sources of flexibility. First, the algorithm self‑adapts its key parameters (distance cutoff, minimum fragment length, scoring thresholds) to the size of the input proteins, thereby maintaining sensitivity for short proteins and avoiding an explosion of fragment candidates for long proteins. Second, instead of enforcing a strict one‑to‑one correspondence between aligned fragment pairs (AFPs), CLeFAPS allows an AFP to be inserted into a one‑to‑many correspondence set, effectively bypassing the traditional position‑conflict check and enabling the capture of genuine structural variations. Third, the extension step that grows initial AFPs into longer aligned regions relies on a vector‑based scoring scheme rather than a pure Euclidean distance metric; by comparing the direction vectors of adjacent fragments, the method remains invariant to rigid‑body transformations and can elongate alignments without being misled by local bends or twists.
A central innovation is the use of Conformational Letters (CL), a discrete alphabet that maps backbone dihedral angles (ϕ, ψ) to 17 pre‑defined symbols. This conversion turns three‑dimensional structures into strings, allowing fast hash‑based matching analogous to sequence alignment while dramatically reducing computational cost and increasing robustness to noise. After an initial CL‑based seed search, the three flexibility mechanisms are applied iteratively to refine the alignment and compute the final superposition.
The authors benchmarked CLeFAPS on two widely used datasets. HOMSTRAD, containing closely related protein pairs, was used to assess alignment accuracy under conditions where high similarity is expected. SABmark, which includes distantly related proteins and provides a fold‑discrimination test, evaluated the algorithm’s ability to distinguish true structural homologs from unrelated decoys. On HOMSTRAD, CLeFAPS achieved an average TM‑score of 0.78 and RMSD of 2.1 Å, comparable to or slightly better than classic rigid‑body methods such as TM‑align and DALI. On the more challenging SABmark set, it obtained an average TM‑score of 0.55 and RMSD of 3.4 Å, with a specificity of 0.93 and sensitivity of 0.89 in the discrimination task, matching or surpassing flexible methods like MUSTANG and FATCAT.
Speed is where CLeFAPS truly shines. The average runtime per protein pair was approximately 0.04 seconds, representing a 1/50 to 1/150 reduction relative to the competing algorithms (TM‑align ~2 s, FATCAT ~3 s). This dramatic acceleration stems from the CL string representation, the automatic scaling of parameters, and the avoidance of exhaustive conflict resolution during AFP insertion.
In summary, CLeFAPS delivers a compelling combination of high alignment quality and ultra‑fast execution. By converting structures to a discrete alphabet, adapting parameters to protein size, allowing many‑to‑one fragment correspondences, and employing vector‑based extension, it captures biologically relevant flexibility without sacrificing speed. The method is poised for integration into large‑scale structural genomics pipelines, rapid homology detection, and multi‑structure comparative analyses, where both accuracy and throughput are critical.
Comments & Academic Discussion
Loading comments...
Leave a Comment