Adaptation through stochastic switching into transient mutators in finite asexual populations
The importance of mutator clones in the adaptive evolution of asexual populations is not fully understood. Here we address this problem by using an ab initio microscopic model of living cells, whose fitness is derived directly from their genomes using a biophysically realistic model of protein folding and interactions in the cytoplasm. The model organisms contain replication controlling genes (DCGs) and genes modeling the mismatch repair (MMR) complexes. We find that adaptation occurs through the transient fixation of a mutator phenotype, regardless of particular perturbations in the fitness landscape. The microscopic pathway of adaptation follows a well-defined set of events: stochastic switching to the mutator phenotype first, then mutation in the MMR complex that hitchhikes with a beneficial mutation in the DCGs, and finally a compensating mutation in the MMR complex returning the population to a non-mutator phenotype. Similarity of these results to reported adaptation events points out to robust universal physical principles of evolutionary adaptation.
💡 Research Summary
The paper tackles a long‑standing question in evolutionary biology: how do mutator clones contribute to the adaptive dynamics of asexual populations? To answer this, the authors construct an “ab initio” microscopic model of living cells in which fitness is not imposed by an abstract function but emerges directly from the physical properties of proteins encoded by the genome. Each model organism carries two functional modules: (i) a replication‑controlling gene (DCG) whose protein product determines the cell’s replication rate, and (ii) a mismatch‑repair (MMR) complex that reduces the genomic mutation rate. The model explicitly computes protein folding free energies, intracellular concentrations, and pairwise binding affinities; these quantities are then translated into a growth rate that serves as the organism’s fitness. By grounding evolution in biophysical reality, the model can capture how a single nucleotide change propagates through protein stability, interaction networks, and ultimately population dynamics.
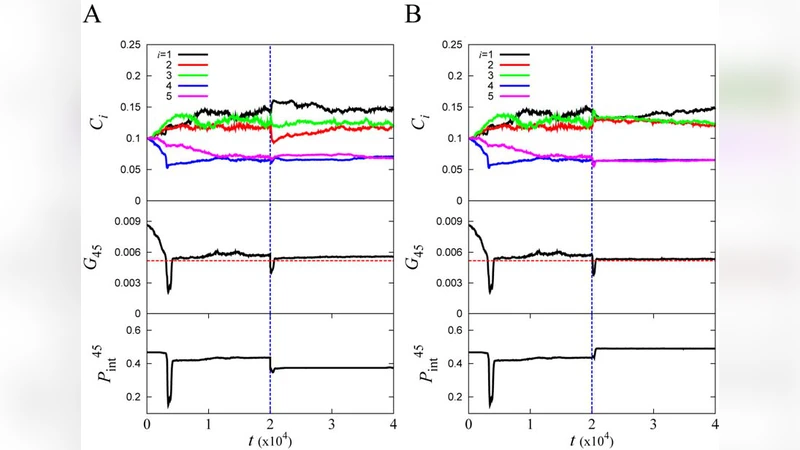
Simulations were performed under a variety of environmental perturbations—nutrient limitation, temperature shifts, and exposure to toxic compounds—to test whether the adaptive pathway depends on the nature of the stress. Remarkably, in every scenario the population followed the same four‑stage trajectory. The first stage is a stochastic switch: random fluctuations in MMR gene expression or protein stability temporarily impair mismatch repair, producing a transient mutator phenotype. During this window the genome‑wide mutation rate spikes by one to two orders of magnitude, generating a flood of new variants. In the second stage a loss‑of‑function mutation in the MMR complex becomes fixed, cementing the mutator state. Crucially, this mutator background dramatically raises the probability that a beneficial mutation will arise in a DCG.
The third stage is a classic hitchhiking event: the beneficial DCG mutation and the MMR mutator allele rise in frequency together, because the fitness advantage conferred by the DCG mutation outweighs the deleterious load of the elevated mutation rate. The population therefore experiences a rapid sweep of a combined genotype (mutator + advantageous DCG). Finally, in the fourth stage a compensatory mutation occurs within the MMR complex, restoring its repair efficiency. This “reversion” eliminates the mutator phenotype, stabilizing the newly adapted genotype at a lower, near‑wild‑type mutation rate.
These findings diverge from traditional mutator models that assume a mutator clone first fixes and then later acquires a beneficial mutation. Here the mutator phenotype is explicitly transient and precedes the fixation of the advantageous allele; the subsequent compensatory change in MMR is essential for the population to exit the high‑mutation regime. Moreover, the stochastic emergence of the mutator does not require an external stress signal; it is an intrinsic property of the replication‑repair system’s biophysical noise. This insight aligns with experimental observations in bacteria such as E. coli and Pseudomonas, where loss‑of‑function mutations in mutS/mutL appear early during adaptation to antibiotics, followed by secondary mutations that restore mismatch repair.
The authors argue that this four‑step pathway reflects a universal physical principle of adaptation: a temporary breach in the fidelity of information transmission (the mutator switch) creates a burst of genetic diversity, which is then sculpted by selection on functional modules, and finally the system self‑corrects to maintain long‑term genomic stability. The model’s predictive power lies in its ability to reproduce quantitative features of laboratory evolution experiments—such as the timing of mutator peaks, the frequency of hitchhiking events, and the eventual decline of mutation rates—without invoking ad‑hoc fitness landscapes.
In conclusion, the study provides a mechanistic, physics‑based framework for understanding how transient mutator states drive rapid adaptation in finite asexual populations. By linking protein‑level biophysics to population‑level evolutionary outcomes, it bridges a critical gap between molecular biology and evolutionary theory. The implications are broad: they inform strategies to curb the emergence of antibiotic resistance (by targeting the mutator window), guide the design of synthetic evolution platforms where controlled mutator phases could accelerate the discovery of novel functions, and suggest that many adaptive processes across biology may be governed by similar transient fidelity‑breakdown mechanisms.
Comments & Academic Discussion
Loading comments...
Leave a Comment