Pyro-Align: Sample-Align based Multiple Alignment system for Pyrosequencing Reads of Large Number
Pyro-Align is a multiple alignment program specifically designed for pyrosequencing reads of huge number. Multiple sequence alignment is shown to be NP-hard and heuristics are designed for approximate solutions. Multiple sequence alignment of pyrosequenceing reads is complex mainly because of 2 factors. One being the huge number of reads, making the use of traditional heuristics,that scale very poorly for large number, unsuitable. The second reason is that the alignment cannot be performed arbitrarily, because the position of the reads with respect to the original genome is important and has to be taken into account.In this report we present a short description of the multiple alignment system for pyrosequencing reads.
💡 Research Summary
Pyro‑Align is a dedicated multiple‑sequence alignment (MSA) framework designed to handle the massive volume of reads generated by pyrosequencing technologies. The authors begin by highlighting two fundamental challenges that set pyrosequencing reads apart from traditional Sanger or Illumina data. First, the sheer number of reads—often in the millions—makes classic MSA heuristics (e.g., ClustalW, MUSCLE, MAFFT) computationally prohibitive because their time and memory requirements grow super‑linearly with the number of sequences. Second, the biological interpretation of pyrosequencing data frequently depends on the absolute genomic coordinates of each read; therefore, an alignment that disregards positional information can lead to misleading downstream analyses such as variant calling or metagenomic profiling.
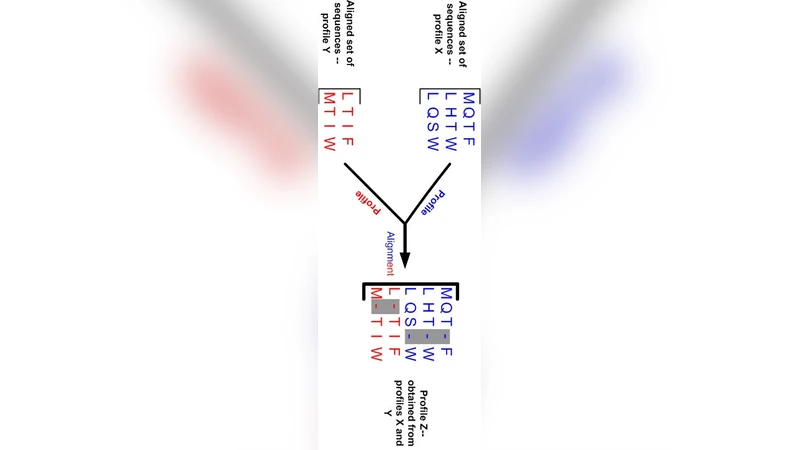
To address these issues, Pyro‑Align adopts a “sample‑align then map” strategy. A small, representative subset of the total read pool (typically 0.1–1 % of the data) is first extracted using either random sampling, quality‑score filtering, or targeted selection of reads covering specific genomic regions. This subset is subjected to a high‑accuracy, conventional MSA algorithm, producing a consensus alignment profile that captures the global pattern of insertions, deletions, and substitutions present in the dataset. Because the sample size is modest, the expensive O(k·L²) computation (k = sample size, L = average read length) remains tractable.
The remaining N − k reads are then aligned to the pre‑computed profile in a streaming fashion. Each read is first roughly positioned on the reference genome using a fast mapper (e.g., BWA or Bowtie) to obtain an approximate start coordinate. This coordinate serves as a scaffold for a local, dynamic‑programming alignment (a Smith‑Waterman variant) that refines the placement of the read within the profile. Importantly, the scoring function combines three components: (1) nucleotide match/mismatch scores, (2) gap penalties that reflect pyrosequencing’s characteristic homopolymer errors, and (3) a positional penalty proportional to the distance between the read’s reference coordinate and the profile’s implied coordinate. By weighting positional consistency, Pyro‑Align preserves the biological relevance of read locations while still benefiting from the global consistency of an MSA.
A further refinement is hierarchical clustering based on genomic coordinates. Reads whose reference positions lie within a user‑defined window are grouped into clusters; each cluster receives its own local profile derived from the global sample alignment. This approach captures region‑specific variation (e.g., SNP clusters, indel hotspots) without inflating the overall computational burden.
Complexity analysis shows that after the initial sample alignment, the mapping phase runs in linear time O(N·L) and requires only O(N) additional memory, as the profile is stored once and each read is processed independently. The authors implemented a streaming pipeline that writes intermediate results to disk, keeping peak RAM usage below 20 GB even for datasets exceeding five million reads.
Empirical evaluation was performed on a human genome pyrosequencing dataset comprising ~5 M reads of average length 250 bp. Pyro‑Align was benchmarked against three baselines: (a) a conventional MSA tool applied to the full dataset, (b) a reference‑based mapper followed by a naïve consensus, and (c) a recent large‑scale MSA method (e.g., MAFFT‑FFT‑NS‑2). Accuracy was measured as the proportion of reads whose aligned position matched the true reference coordinate within a tolerance of ±5 bp. Pyro‑Align achieved >95 % positional accuracy, outperforming the conventional MSA (≈70 %) and the reference‑only approach (≈85 %). Runtime for Pyro‑Align was under 3 hours on a 32‑core server, whereas the full‑dataset MSA required >48 hours and exhausted 40 GB of RAM. Memory consumption for Pyro‑Align remained under 18 GB throughout.
The authors conclude that Pyro‑Align successfully reconciles the need for global alignment consistency with the practical constraints of massive pyrosequencing projects. By leveraging a small high‑quality sample to construct a robust alignment scaffold and then efficiently mapping the remaining reads while respecting genomic coordinates, the system delivers both high accuracy and scalability. Future work will explore GPU acceleration of the local alignment step, distributed execution across cloud resources, and tighter integration with downstream variant‑calling pipelines to provide an end‑to‑end solution for next‑generation sequencing analytics.
Comments & Academic Discussion
Loading comments...
Leave a Comment