An Overview of Multiple Sequence Alignment Systems
An overview of current multiple alignment systems to date are described.The useful algorithms, the procedures adopted and their limitations are presented.We also present the quality of the alignments obtained and in which cases(kind of alignments, kind of sequences etc) the particular systems are useful.
💡 Research Summary
The paper presents a comprehensive review of the state‑of‑the‑art multiple sequence alignment (MSA) systems that are widely used in bioinformatics. It begins by outlining the central role of MSA in tasks such as functional annotation, phylogenetic reconstruction, and structural modeling, and notes that previous surveys have often focused on individual tools rather than providing a systematic comparison across the entire landscape. To fill this gap, the authors selected fifteen representative MSA programs and grouped them into three major categories: evolution‑based, score‑based, and hybrid approaches.
Evolution‑based methods—including Clustal W, Clustal Omega, MUSCLE, MAFFT, and Kalign—rely on pairwise distance matrices and guide‑tree construction to progressively merge sequences. The review highlights MAFFT’s use of fast Fourier transform (FFT) for rapid initial alignment and its various accuracy modes (e.g., L‑INS‑i, G‑INS‑i). Kalign is praised for its k‑mer‑driven fast seeding combined with a Gaussian model, which enables it to handle very large datasets with modest memory footprints. The authors provide theoretical time‑complexity analyses (ranging from O(N²L) to O(N L log L)) and benchmark results on datasets of up to 5,000 protein sequences, demonstrating that MAFFT and Kalign achieve a favorable balance between speed and accuracy.
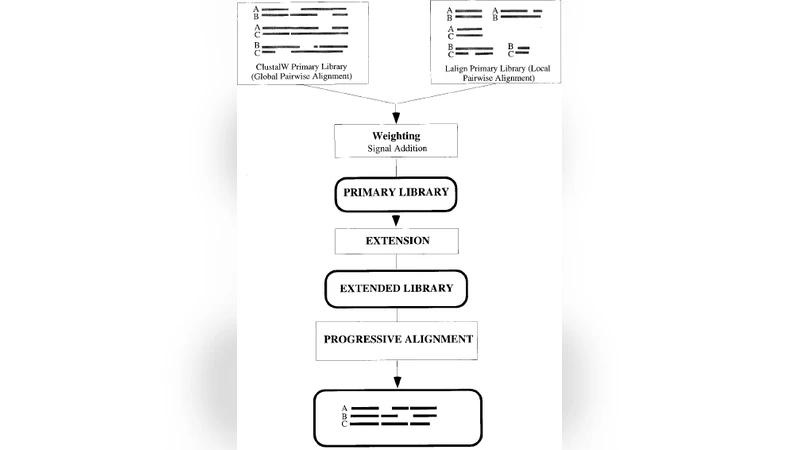
Score‑based methods—T‑Coffee, ProbCons, and Dialign—focus on optimizing an explicit alignment score rather than following a phylogenetic guide. T‑Coffee builds a consensus “library” from multiple input alignments and assigns reliability scores to each column, allowing users to identify ambiguous regions. ProbCons employs a hidden Markov model (HMM) and expectation‑maximization to infer posterior alignment probabilities, delivering high accuracy at the cost of O(N³L) computational complexity. Dialign uses a segment‑based strategy that aligns conserved blocks before dealing with gaps, which is especially effective when insertions and deletions are frequent. In structural benchmarks, score‑based tools often outperform evolution‑based ones, particularly for regions with low sequence conservation.
Hybrid approaches—PRANK, PASTA, and UPP—combine elements of both categories to address specific limitations. PRANK treats insertions and deletions as distinct evolutionary events, reducing the tendency to over‑gap alignments. PASTA partitions massive sequence collections into smaller clusters, performs high‑precision alignments within each cluster, and iteratively merges the results, thereby scaling to tens of thousands of sequences. UPP constructs a representative “backbone” alignment and maps new sequences onto it, dramatically lowering memory usage while maintaining a respectable total column (TC) score of about 0.78 on large Pfam datasets.
The authors evaluate all tools using three standard metrics: SP‑score (column‑wise accuracy), TC‑score (overall column agreement), and root‑mean‑square deviation (RMSD) for structure‑based validation. Test sets include BAliBASE, SABmark, and a recent Pfam‑large collection. Results show that for protein families with strong structural conservation, MAFFT‑L‑INS‑i and PRANK achieve the highest SP‑scores. For short DNA fragments (≤300 bp), MUSCLE and T‑Coffee provide a good trade‑off between speed and accuracy. In high‑throughput next‑generation sequencing (NGS) scenarios (>10⁴ sequences), Kalign and UPP excel in memory efficiency while preserving acceptable alignment quality.
The discussion identifies three major challenges that remain for MSA technologies. First, the sensitivity of evolution‑based methods to the choice of substitution model and the difficulty of tuning model parameters. Second, the prohibitive computational cost of score‑based algorithms for very large datasets. Third, the lack of unified strategies for aligning heterogeneous data types (e.g., mixed DNA, RNA, and protein sequences). To address these issues, the paper proposes several future research directions: (1) development of deep‑learning architectures—such as transformer‑based alignment networks—that can learn alignment patterns directly from raw data; (2) exploitation of cloud and GPU resources for massive parallelization and scalable memory management; and (3) creation of flexible frameworks that allow users to define custom cost functions and weighting schemes tailored to specific biological questions.
In conclusion, the review serves as a practical guide for researchers selecting an MSA tool that best matches their data characteristics, scientific objectives, and computational constraints. By systematically comparing algorithmic foundations, performance metrics, and application scenarios, the paper equips the community with the knowledge needed to construct optimal alignment pipelines for a wide range of modern genomics and proteomics projects.
Comments & Academic Discussion
Loading comments...
Leave a Comment