Computer algebra in systems biology
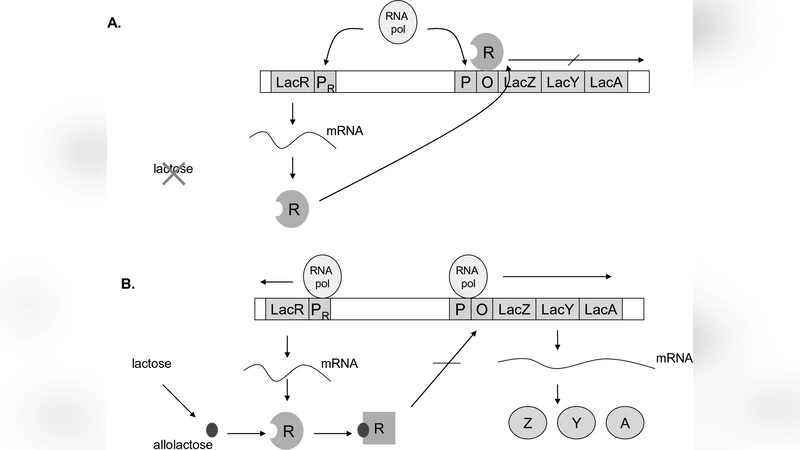
Systems biology focuses on the study of entire biological systems rather than on their individual components. With the emergence of high-throughput data generation technologies for molecular biology and the development of advanced mathematical modeling techniques, this field promises to provide important new insights. At the same time, with the availability of increasingly powerful computers, computer algebra has developed into a useful tool for many applications. This article illustrates the use of computer algebra in systems biology by way of a well-known gene regulatory network, the Lac Operon in the bacterium E. coli.
💡 Research Summary
Systems biology aims to understand the behavior of entire biological networks rather than isolated components. The rapid expansion of high‑throughput technologies—such as next‑generation sequencing, microarrays, and mass spectrometry—has generated massive quantitative datasets that enable the construction of detailed mathematical models. However, these models are typically nonlinear, high‑dimensional, and contain many uncertain parameters, making purely numerical approaches insufficient for a global analysis of system behavior.
In this paper the authors propose computer algebra, i.e., symbolic computation, as a complementary tool for systems biology. Symbolic methods such as Gröbner basis computation, elimination theory, and normal‑form transformations allow one to manipulate polynomial equations exactly, to eliminate variables, and to derive explicit conditions on parameters that guarantee the existence and multiplicity of steady‑state solutions. By working with exact expressions rather than floating‑point approximations, one avoids cumulative numerical errors and obtains rigorous statements about the structure of the solution space.
The authors illustrate the approach with the classic Lac Operon regulatory network of Escherichia coli. The operon controls lactose utilization through a feedback loop involving transcription, translation, and inducer binding. Traditional studies have relied on numerical simulations to locate bistable regimes, but these simulations explore only a limited set of parameter values. Here the operon is modeled by three coupled nonlinear ordinary differential equations. Setting the time derivatives to zero yields a system of polynomial equations in the concentrations of mRNA, repressor protein, and intracellular lactose. Using a computer algebra system (e.g., Maple, Mathematica, Singular), the authors compute a Gröbner basis that systematically eliminates variables, ultimately reducing the problem to a single polynomial whose coefficients depend on biologically relevant parameters such as transcription rates, degradation constants, and binding affinities.
Analyzing the sign and multiplicity of the roots of this reduced polynomial provides explicit algebraic inequalities that delineate regions of monostability, bistability, and even tristability in parameter space. In particular, the authors derive a critical curve relating the transcriptional repression strength to the inducer binding affinity, showing precisely where a saddle‑node bifurcation occurs. This symbolic result guides experimental design: by tuning specific kinetic constants, researchers can induce or suppress the desired switching behavior.
The paper emphasizes that symbolic analysis and numerical simulation are synergistic. Symbolic computation identifies the global structure of the parameter space, dramatically narrowing the region that must be explored numerically. Within the identified region, time‑course simulations can then quantify dynamic properties such as switching times, hysteresis width, and robustness to noise. The authors present a hybrid workflow: first perform algebraic elimination to obtain parameter constraints, then conduct targeted numerical experiments to validate and refine the predictions. This approach reduces computational cost while preserving analytical rigor.
Finally, the authors discuss current limitations and future directions. Gröbner basis calculations scale poorly with the number of variables and the degree of the polynomials, leading to combinatorial explosion for large networks. Emerging strategies—modular decomposition, modular Gröbner bases, parallel algorithms, and hybrid symbolic‑numeric methods—are being developed to mitigate this bottleneck. Extending symbolic techniques to stochastic models, hybrid discrete‑continuous systems, and parameter‑identification problems remains an open research frontier. As these methodological advances mature, computer algebra is poised to become a central pillar of systems biology, enabling rigorous analysis of large‑scale gene regulatory networks, metabolic pathways, and synthetic biology designs.
Comments & Academic Discussion
Loading comments...
Leave a Comment