Introduction to protein folding for physicists
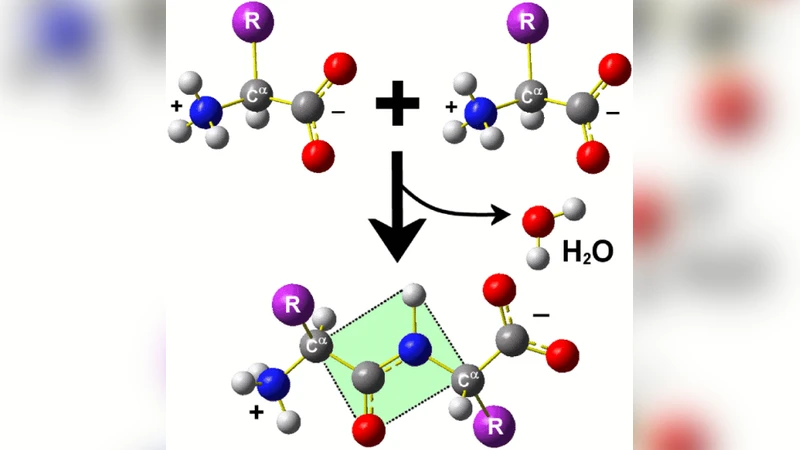
The prediction of the three-dimensional native structure of proteins from the knowledge of their amino acid sequence, known as the protein folding problem, is one of the most important yet unsolved issues of modern science. Since the conformational behaviour of flexible molecules is nothing more than a complex physical problem, increasingly more physicists are moving into the study of protein systems, bringing with them powerful mathematical and computational tools, as well as the sharp intuition and deep images inherent to the physics discipline. This work attempts to facilitate the first steps of such a transition. In order to achieve this goal, we provide an exhaustive account of the reasons underlying the protein folding problem enormous relevance and summarize the present-day status of the methods aimed to solving it. We also provide an introduction to the particular structure of these biological heteropolymers, and we physically define the problem stating the assumptions behind this (commonly implicit) definition. Finally, we review the ‘special flavor’ of statistical mechanics that is typically used to study the astronomically large phase spaces of macromolecules. Throughout the whole work, much material that is found scattered in the literature has been put together here to improve comprehension and to serve as a handy reference.
💡 Research Summary
The paper serves as a bridge for physicists entering the field of protein folding, offering a comprehensive overview of the problem’s significance, the unique physical characteristics of proteins, and the methodological landscape. It begins by emphasizing that the three‑dimensional native structure of a protein, dictated solely by its amino‑acid sequence, underlies all biological function, making the prediction of this structure a central unsolved challenge. Unlike homogeneous synthetic polymers, proteins are heteropolymers with a rich variety of side‑chain chemistries, directional hydrogen bonds, hydrophobic effects, and steric constraints, which together generate a highly rugged free‑energy landscape.
The authors formally define the folding problem as the search for the global minimum of the free‑energy surface under fixed temperature, pressure, and solvent conditions—assumptions that are often implicit in the literature but are explicitly examined here. They then present the statistical‑mechanical framework that has become standard for tackling the astronomically large conformational space. Central to this framework is the “folding funnel” concept, which rationalizes how a protein can efficiently navigate toward its native basin despite the combinatorial explosion of possible configurations. Classical sampling techniques such as Monte Carlo and molecular dynamics are described, together with advanced algorithms—replica‑exchange, metadynamics, Gaussian‑accelerated MD—that enhance exploration of high‑energy barriers.
The review proceeds to discuss coarse‑grained models (HP lattice, Go‑type potentials) and more detailed all‑atom force fields, highlighting their respective strengths and limitations. A dedicated section addresses the recent surge of deep‑learning approaches, especially AlphaFold and RoseTTAFold, explaining how data‑driven predictions complement physics‑based simulations by providing high‑accuracy structural priors and by informing force‑field parameterization.
Experimental validation is treated as an essential component: X‑ray crystallography, NMR spectroscopy, and cryo‑electron microscopy supply structural benchmarks and dynamical information that are used to test and refine computational models. The authors stress the bidirectional flow of information—experiments guide simulations, while simulations predict transient intermediates that are difficult to capture experimentally.
Finally, the paper outlines current bottlenecks: limited force‑field accuracy, insufficient sampling of micro‑ to millisecond timescales, and prohibitive computational cost. Prospective solutions include multiscale modeling that couples coarse‑grained and atomistic descriptions, QM/MM hybrid methods for critical regions, and the integration of large‑scale machine‑learning models with traditional statistical‑mechanical techniques. In conclusion, the authors provide a step‑by‑step roadmap for physicists: acquire a solid grounding in protein chemistry, master statistical‑mechanical tools, become proficient with modern simulation software, and engage with experimental data and AI‑based predictors. This synthesis of theory, computation, and experiment is presented as the most promising path toward solving the protein folding problem.
Comments & Academic Discussion
Loading comments...
Leave a Comment