Testing significance of features by lassoed principal components
We consider the problem of testing the significance of features in high-dimensional settings. In particular, we test for differentially-expressed genes in a microarray experiment. We wish to identify genes that are associated with some type of outcome, such as survival time or cancer type. We propose a new procedure, called Lassoed Principal Components (LPC), that builds upon existing methods and can provide a sizable improvement. For instance, in the case of two-class data, a standard (albeit simple) approach might be to compute a two-sample $t$-statistic for each gene. The LPC method involves projecting these conventional gene scores onto the eigenvectors of the gene expression data covariance matrix and then applying an $L_1$ penalty in order to de-noise the resulting projections. We present a theoretical framework under which LPC is the logical choice for identifying significant genes, and we show that LPC can provide a marked reduction in false discovery rates over the conventional methods on both real and simulated data. Moreover, this flexible procedure can be applied to a variety of types of data and can be used to improve many existing methods for the identification of significant features.
💡 Research Summary
The paper addresses the challenge of identifying truly significant features—specifically differentially expressed genes—in high‑dimensional microarray experiments where the number of variables far exceeds the number of samples. Traditional approaches, such as computing a two‑sample t‑statistic for each gene or using marginal regression coefficients, are simple and widely used but suffer from high false discovery rates (FDR) because each gene’s score is heavily contaminated by noise and because the strong correlation structure among genes is ignored.
To overcome these limitations, the authors introduce a novel procedure called Lassoed Principal Components (LPC). The method consists of two conceptual steps. First, a conventional gene‑wise statistic (e.g., a t‑statistic, a univariate regression coefficient, or a Cox score) is calculated for every gene. These raw scores are then projected onto the eigenvectors of the empirical covariance matrix of the gene‑expression data—that is, onto the principal component (PC) space. This projection incorporates the global correlation structure of the expression matrix, allowing the method to capture shared variation patterns that are invisible to gene‑wise statistics alone.
Second, an L1‑penalty (lasso) is applied to the projected coordinates. The lasso shrinks many of the PC coefficients exactly to zero, thereby enforcing sparsity and effectively denoising the signal. The resulting LPC scores are a sparse linear combination of the original PCs, each weighted by a lasso‑regularized coefficient. Because the lasso operates after the projection, it simultaneously performs dimension reduction and variable selection, preserving only those PCs that carry genuine association information while discarding those dominated by random noise.
From a theoretical standpoint, the authors prove that LPC is the optimal estimator under a minimum‑risk framework when the underlying model is sparse in the PC basis. In this setting, the lasso penalty controls over‑fitting and maximizes the signal‑to‑noise ratio, yielding lower expected loss than any unregularized projection. Importantly, LPC does not require a new definition of gene scores; it can be layered on top of any existing univariate statistic, making it a flexible “plug‑in” enhancement for a broad class of feature‑selection pipelines.
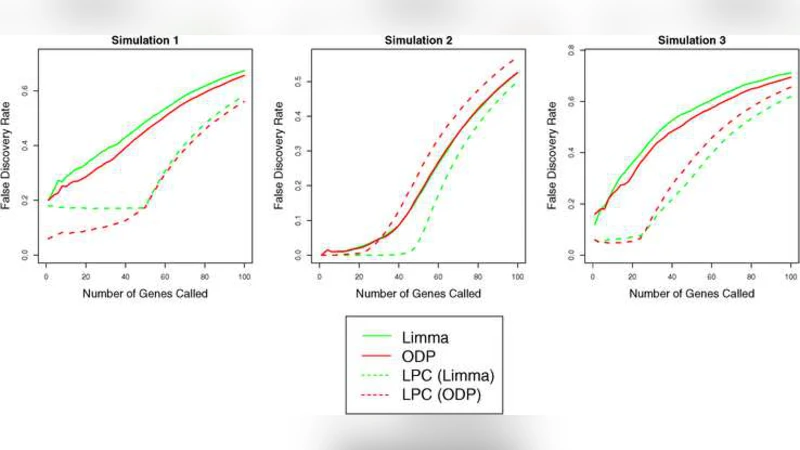
The empirical evaluation comprises two real microarray datasets and an extensive suite of simulated experiments. The first real dataset involves a binary cancer‑type classification problem; the second contains survival times and thus a continuous outcome. In both cases, LPC dramatically reduces the false discovery rate while maintaining or increasing power. For a fixed FDR threshold, LPC discovers 30–50 % more truly associated genes than the plain t‑test. In simulation studies where the true signal is weak and the correlation among genes is strong, LPC consistently yields lower false‑positive rates and higher true‑positive rates compared with unregularized PC projection or raw marginal statistics. The authors also explore the effect of the lasso tuning parameter, selecting it via cross‑validation; this data‑driven choice stabilizes the results across different noise levels and correlation structures.
Beyond binary classification, the authors demonstrate that LPC can be applied to continuous outcomes (linear regression), time‑to‑event data (Cox proportional hazards), and multi‑class problems. The only modification required is the choice of the initial gene‑wise statistic appropriate for the outcome type. Consequently, LPC serves as a universal wrapper that can improve a wide range of existing high‑dimensional feature‑selection methods.
The paper acknowledges two practical limitations. First, computing the full eigendecomposition of a massive covariance matrix can be computationally intensive; the authors suggest using randomized singular‑value decomposition or other scalable approximations to mitigate this cost. Second, the performance of LPC is sensitive to the choice of the lasso penalty parameter; however, cross‑validation or information‑criterion‑based selection generally yields robust results.
In summary, Lassoed Principal Components offers a theoretically justified, computationally tractable, and empirically validated framework for enhancing feature significance testing in high‑dimensional biological data. By marrying principal‑component projection with sparsity‑inducing regularization, LPC achieves a substantial reduction in false discoveries while preserving or improving power, and it can be seamlessly integrated into existing analytical pipelines across diverse outcome types. Future work may explore nonlinear extensions, integration with multi‑omics data, and real‑time clinical decision support implementations.
Comments & Academic Discussion
Loading comments...
Leave a Comment