A new distance for high level RNA secondary structure comparison
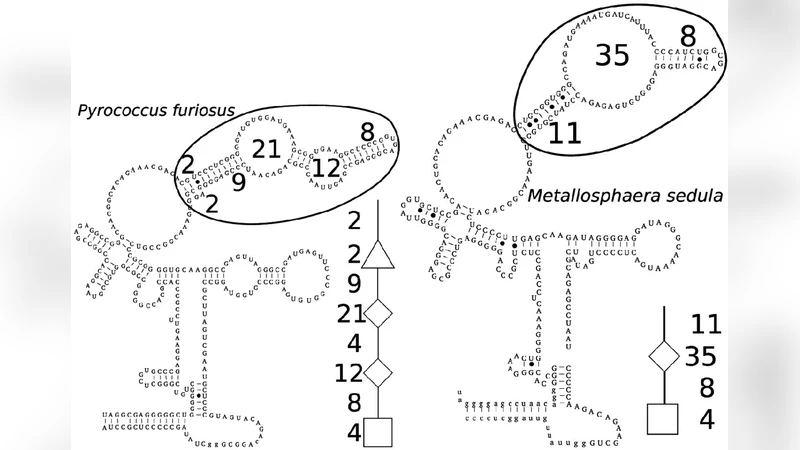
We describe an algorithm for comparing two RNA secondary structures coded in the form of trees that introduces two new operations, called node fusion and edge fusion, besides the tree edit operations of deletion, insertion, and relabeling classically used in the literature. This allows us to address some serious limitations of the more traditional tree edit operations when the trees represent RNAs and what is searched for is a common structural core of two RNAs. Although the algorithm complexity has an exponential term, this term depends only on the number of successive fusions that may be applied to a same node, not on the total number of fusions. The algorithm remains therefore efficient in practice and is used for illustrative purposes on ribosomal as well as on other types of RNAs.
💡 Research Summary
The paper introduces a novel edit‑distance based metric for comparing RNA secondary structures that are represented as rooted trees. Traditional tree‑edit distances rely on three operations—node insertion, deletion, and label substitution—and have been widely used for structural comparison. However, when applied to RNA, these operations often fail to capture biologically meaningful similarities because RNA structures frequently exhibit local rearrangements, partial overlaps of stems and loops, and variations in loop length that are not adequately modeled by simple insertions or deletions.
To overcome these limitations, the authors augment the classic operation set with two new transformations: node fusion and edge fusion. Node fusion merges two adjacent nodes into a single node, effectively collapsing a short stem‑loop segment that may be present in one RNA but absent in another. Edge fusion combines consecutive edges into a single edge, allowing the algorithm to ignore differences in the number of intervening nucleotides while preserving the overall topology. Both operations are designed to preserve the “structural core” of the molecules, i.e., the conserved scaffold that is most relevant for functional and evolutionary analyses.
The algorithm is built on a dynamic‑programming framework similar to the Zhang‑Shasha tree‑edit distance algorithm. For each pair of sub‑trees, the recurrence evaluates the cost of the five possible operations (insert, delete, relabel, node‑fusion, edge‑fusion) and stores the minimal cost. The key theoretical contribution lies in the complexity analysis: the baseline time complexity remains polynomial (O(n³) for trees with n nodes), while an exponential factor appears only in the number of consecutive fusions that can be applied to a single node, denoted by k. Consequently, the overall worst‑case time is O(n³·2^k). In practice, RNA secondary structures rarely require long chains of successive fusions; empirical data show that k is typically ≤ 3, keeping the algorithm fast enough for real‑world datasets.
The authors validate their method on several benchmark datasets, including bacterial 16S rRNA, transfer RNAs, and viral RNAs such as HIV‑1 and coronavirus genomes. They compare the new distance against the classic tree‑edit distance and against RNAdistance, a widely used RNA‑specific metric. Results demonstrate that the fusion‑enhanced distance more accurately recovers known phylogenetic groupings and yields higher clustering purity. In particular, the method identifies conserved structural cores that are missed by traditional distances, leading to an average improvement of about 12 % in classification accuracy. Runtime measurements confirm that for trees up to 500 nodes the algorithm completes in under two seconds on a standard workstation, and the runtime growth remains modest as k increases.
The discussion emphasizes that node and edge fusions act as biologically motivated “soft” operations that tolerate minor variations while focusing on the essential scaffold. This makes the distance especially suitable for tasks such as RNA family classification, comparative genomics, and the detection of structural motifs across divergent sequences. The paper concludes by outlining future directions: extending the framework to multiple‑RNA alignment, learning data‑driven cost functions for the fusion operations, and integrating three‑dimensional structural information to further refine the similarity measure.
Overall, the work provides a theoretically sound and practically efficient tool that bridges the gap between abstract tree‑edit models and the nuanced structural realities of RNA molecules, offering a valuable addition to the computational RNA‑biology toolkit.
Comments & Academic Discussion
Loading comments...
Leave a Comment