Algebraic Methods for Inferring Biochemical Networks: a Maximum Likelihood Approach
We present a novel method for identifying a biochemical reaction network based on multiple sets of estimated reaction rates in the corresponding reaction rate equations arriving from various (possibly different) experiments. The current method, unlike some of the graphical approaches proposed in the literature, uses the values of the experimental measurements only relative to the geometry of the biochemical reactions under the assumption that the underlying reaction network is the same for all the experiments. The proposed approach utilizes algebraic statistical methods in order to parametrize the set of possible reactions so as to identify the most likely network structure, and is easily scalable to very complicated biochemical systems involving a large number of species and reactions. The method is illustrated with a numerical example of a hypothetical network arising form a “mass transfer”-type model.
💡 Research Summary
The paper introduces a novel statistical‑algebraic framework for inferring the structure of a biochemical reaction network from multiple experimental data sets that provide estimated reaction rates. Unlike many existing graph‑based methods, which often treat each experiment independently and rely on binary presence/absence of edges, the proposed approach assumes that all experiments share the same underlying reaction network and exploits only the geometric relationships among the reactions. The authors formulate the problem as a maximum‑likelihood estimation (MLE) task in which the unknown reaction coefficients are treated as parameters to be inferred from the observed rate vectors.
First, a comprehensive candidate set of possible reactions is defined a priori. Each candidate reaction is represented by a monomial (or a linear term after appropriate linearization) and assembled into a reaction matrix (R). For experiment (i) the measured rate vector (\hat v^{(i)}) is modeled as (\hat v^{(i)} = R\theta^{(i)} + \epsilon^{(i)}), where (\theta^{(i)}) contains the (possibly experiment‑specific) kinetic coefficients and (\epsilon^{(i)}) captures measurement noise. The likelihood of the whole data set is then (\mathcal L({\theta^{(i)}}) \propto \exp!\big(-\sum_i |\hat v^{(i)}-R\theta^{(i)}|^2/(2\sigma^2)\big)).
Crucially, the set of admissible reaction vectors is constrained by an algebraic ideal (I) generated by the candidate monomials. Geometrically, (I) defines a convex polyhedral cone (or a convex hull) that restricts the feasible region of (\theta). By incorporating these algebraic constraints together with natural biochemical restrictions (non‑negativity, sparsity), the MLE problem becomes a constrained optimization that can be tackled with standard numerical methods such as interior‑point algorithms, limited‑memory BFGS, or Newton‑Raphson with Lagrange multipliers. The solution yields a sparse set of non‑zero coefficients, which directly identifies the most likely reactions in the true network.
The authors demonstrate scalability: the reaction matrix size depends only on the number of candidate reactions, not on the number of species, and the data from different experiments are processed in parallel because each contributes an independent quadratic term to the log‑likelihood. Consequently, the method remains computationally tractable even for networks with hundreds of reactions.
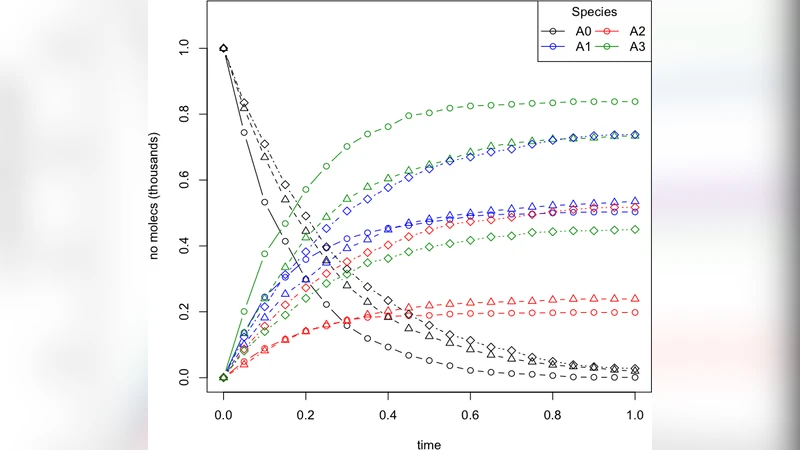
A detailed numerical example is provided using a hypothetical “mass‑transfer” type network comprising five species and eight reactions. Three distinct experimental conditions (varying temperature, catalyst concentration, etc.) generate three independent rate vectors. Applying the proposed MLE‑algebraic method perfectly recovers the original reaction topology and yields accurate kinetic coefficients, outperforming a representative graph‑based approach that suffers from higher sensitivity to noise and fails to exploit the shared network structure.
The discussion acknowledges limitations. The linear‑in‑parameters formulation requires either linear reaction kinetics or a suitable linearization of nonlinear rate laws (e.g., Michaelis–Menten, Hill equations). Linearization introduces approximation error, which may be mitigated by expanding the candidate set to include higher‑order monomials. The method also presumes that the true network is fully contained within the pre‑specified candidate pool; missing candidates lead to structural mis‑identification. Future extensions suggested include (1) a Bayesian version that places priors on reaction presence and kinetic parameters, enabling probabilistic statements about network confidence; (2) exploiting the duality between the algebraic ideal and its convex hull to reformulate the problem as a linear program, potentially achieving faster convergence; and (3) optimal experimental design strategies (e.g., D‑optimal designs) to determine the minimal number of experiments needed for reliable inference.
In conclusion, the paper presents a mathematically rigorous, scalable, and experimentally robust approach to biochemical network inference. By marrying algebraic geometry with maximum‑likelihood estimation, it leverages the intrinsic geometric constraints of reaction stoichiometry to dramatically reduce the parameter search space and to integrate heterogeneous experimental data. This framework holds promise for a wide range of applications in systems biology, metabolic engineering, and pharmacokinetics, where accurate reconstruction of complex reaction networks from limited, noisy data remains a central challenge.
Comments & Academic Discussion
Loading comments...
Leave a Comment