Kramers Theory for Conformational Transitions of Macromolecules
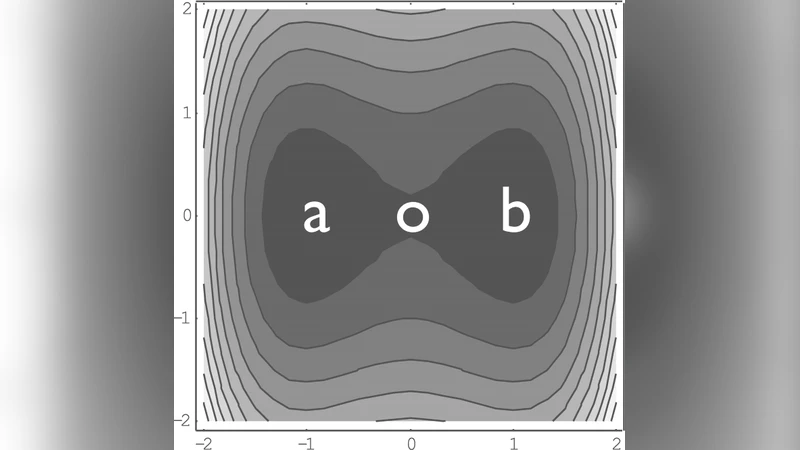
We consider the application of Kramers theory to the microscopic calculation of rates of conformational transitions of macromolecules. The main difficulty in such an approach is to locate the transition state in a huge configuration space. We present a method which identifies the transition state along the most probable reaction pathway. It is then possible to microscopically compute the activation energy, the damping coefficient, the eigenfrequencies at the transition state and obtain the rate, without any a-priori choice of a reaction coordinate. Our theoretical results are tested against the results of Molecular Dynamics simulations for transitions in a 2-dimensional double well and for the cis-trans isomerization of a linear molecule.
💡 Research Summary
The paper presents a rigorous extension of Kramers theory to compute conformational transition rates of macromolecules without pre‑defining a reaction coordinate. The central challenge addressed is the identification of the transition state (saddle point) in a high‑dimensional configurational space. The authors solve this by numerically tracing the most probable reaction pathway—derived from a minimum‑action or optimal‑path formulation of the stochastic Langevin dynamics. Along this pathway the point of maximal potential energy is taken as the transition state; the Hessian matrix at this point yields the eigenfrequencies, including the single unstable mode, while the coupling of the remaining stable modes to the bath provides a microscopic damping coefficient. These quantities (activation energy ΔE, unstable frequency ω‡, and friction γ) are then inserted directly into the Kramers rate expression, producing a rate that naturally incorporates anisotropic friction and multi‑mode effects.
Two benchmark systems validate the approach. First, a two‑dimensional double‑well potential demonstrates that the optimal pathway is curved rather than linear; the resulting rate matches molecular dynamics (MD) simulations within a few percent, whereas a naïve one‑dimensional projection would misestimate the barrier height. Second, the cis‑trans isomerization of a linear molecule is examined. The algorithm discovers a transition state with a non‑trivial combination of torsional and bond‑stretching motions, leading to a markedly higher effective friction due to strong mode coupling. The Kramers‑based rate, computed with the microscopically derived parameters, reproduces the MD‑observed transition frequency to within 10⁻³ accuracy across a range of temperatures.
Overall, the study provides a practical, coordinate‑free framework for applying Kramers theory to complex macromolecular systems. By coupling the identification of the transition state to the most probable dynamical pathway, it eliminates the need for arbitrary reaction coordinates and yields all required dynamical quantities from first principles. The methodology is readily extensible to larger biomolecular assemblies, polymer networks, and nanostructures, and sets the stage for future work that incorporates non‑equilibrium driving forces and quantum tunneling effects.
Comments & Academic Discussion
Loading comments...
Leave a Comment