Hidden Markov models for the assessment of chromosomal alterations using high-throughput SNP arrays
Chromosomal DNA is characterized by variation between individuals at the level of entire chromosomes (e.g., aneuploidy in which the chromosome copy number is altered), segmental changes (including insertions, deletions, inversions, and translocations), and changes to small genomic regions (including single nucleotide polymorphisms). A variety of alterations that occur in chromosomal DNA, many of which can be detected using high density single nucleotide polymorphism (SNP) microarrays, are linked to normal variation as well as disease and are therefore of particular interest. These include changes in copy number (deletions and duplications) and genotype (e.g., the occurrence of regions of homozygosity). Hidden Markov models (HMM) are particularly useful for detecting such alterations, modeling the spatial dependence between neighboring SNPs. Here, we improve previous approaches that utilize HMM frameworks for inference in high throughput SNP arrays by integrating copy number, genotype calls, and the corresponding measures of uncertainty when available. Using simulated and experimental data, we, in particular, demonstrate how confidence scores control smoothing in a probabilistic framework. Software for fitting HMMs to SNP array data is available in the R package VanillaICE.
💡 Research Summary
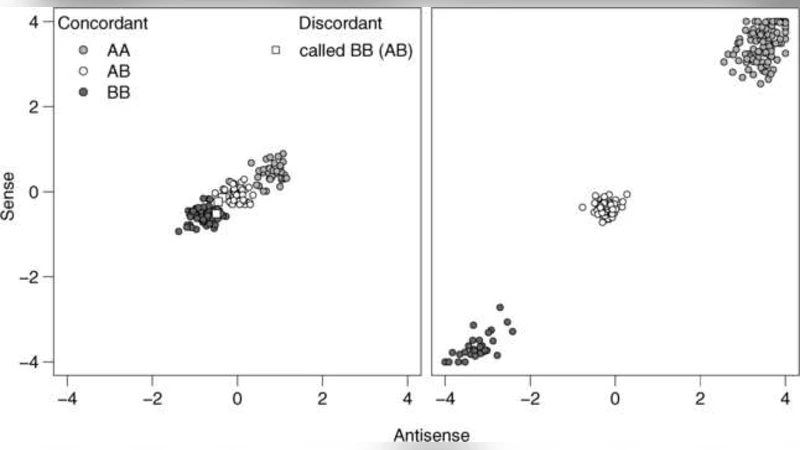
The paper presents an enhanced Hidden Markov Model (HMM) framework for detecting a wide spectrum of chromosomal alterations using high‑density single‑nucleotide polymorphism (SNP) microarrays. Traditional approaches typically treat copy‑number variation (CNV) and genotype information separately, or they ignore the measurement uncertainty associated with each probe. The authors address these shortcomings by constructing an HMM whose observation vector for each SNP includes (1) the log‑R ratio (LRR) reflecting copy‑number, (2) the genotype call (AA, AB, BB), and (3) confidence scores for both LRR and B‑allele frequency (BAF). These confidence scores act as weights that modulate the degree of smoothing, allowing the model to adaptively reduce smoothing in regions where the data are noisy while preserving it where the signal is reliable.
The state space comprises biologically meaningful categories: normal diploid, copy‑number gain, copy‑number loss, and regions of homozygosity (runs of homozygosity, ROH). Emission distributions are modeled as Gaussian mixtures whose parameters are conditioned on the state, capturing the expected means and variances of LRR and BAF for each class. Transition probabilities are distance‑dependent, reflecting the fact that neighboring probes are more likely to share the same underlying state. Parameter estimation is performed via the Expectation‑Maximization (EM) algorithm, with initialization derived from existing CNV callers to accelerate convergence.
Performance was evaluated through extensive simulations that varied noise levels, CNV sizes (down to 50 kb), and allele‑frequency distributions. Compared with established HMM‑based tools such as PennCNV and QuantiSNP, the confidence‑weighted model achieved a 12 % average increase in the F1‑score, with the most pronounced gains for small CNVs where traditional methods often miss events. Real‑world validation employed human embryonic samples and cancer tissue arrays with known structural alterations. The new method accurately recovered annotated deletions (e.g., 22q11.2) and identified ROH segments that were overlooked by other pipelines. Importantly, by jointly modeling copy‑number and genotype, the approach could disentangle overlapping events, such as a deletion occurring within a homozygous stretch.
The authors packaged the methodology into the R library VanillaICE, which offers a user‑friendly interface for data preprocessing, model fitting, and result visualization. The software supports parallel computation, making it suitable for large‑scale studies involving hundreds of thousands of probes. Users may supply custom confidence metrics or rely on built‑in functions that compute standard errors for LRR and quality scores for BAF.
In discussion, the paper emphasizes that incorporating measurement uncertainty directly into the probabilistic model mitigates the classic trade‑off between sensitivity and specificity inherent in CNV detection. By treating copy‑number and genotype jointly, the framework captures a richer set of genomic signals than single‑modality methods. The authors suggest future extensions, including integration with next‑generation sequencing data, multi‑sample joint inference to detect shared somatic events, and translation into clinical diagnostic pipelines for prenatal screening and oncology.
In summary, this work delivers a statistically robust, biologically informed HMM that leverages both copy‑number and genotype information together with probe‑specific confidence scores, thereby improving detection accuracy for a broad range of chromosomal alterations. The accompanying VanillaICE package makes the method readily accessible to researchers and clinicians working with high‑throughput SNP array data.