Studies on the Origin and Evolution of Codon Bias
Background: There is a 3-fold redundancy in the Genetic Code; most amino acids are encoded by more than one codon. These synonymous codons are not used equally; there is a Codon Usage Bias (CUB). This article will provide novel information about the origin and evolution of this bias. Results: Codon Usage Bias (CUB, defined here as deviation from equal usage of synonymous codons) was studied in 113 species. The average CUB was 29.3 +/- 1.1% (S.E.M, n=113) of the theoretical maximum and declined progressively with evolution and increasing genome complexity. A Pan-Genomic Codon Usage Frequency (CUF) Table was constructed to describe genome-wide relationships among codons. Significant correlations were found between the number of synonymous codons and (i) the frequency of the respective amino acids (ii) the size of CUB. Numerous, statistically highly significant, internal correlations were found among codons and the nucleic acids they comprise. These strong correlations made it possible to predict missing synonymous codons (wobble bases) reliably from the remaining codons or codon residues. Conclusions: The results put the concept of “codon bias” into a novel perspective. The internal connectivity of codons indicates that all synonymous codons might be integrated parts of the Genetic Code with equal importance in maintaining its functional integrity.
💡 Research Summary
The manuscript “Studies on the Origin and Evolution of Codon Bias” presents a comprehensive quantitative and qualitative analysis of codon usage bias (CUB) across 113 diverse organisms, ranging from bacteria and archaea to vertebrates. The author constructs a pan‑genomic Codon Usage Frequency (CUF) table by aggregating codon counts from the NCBI Codon Usage Database, thereby creating a single reference that represents roughly 2.88 × 10^11 codons.
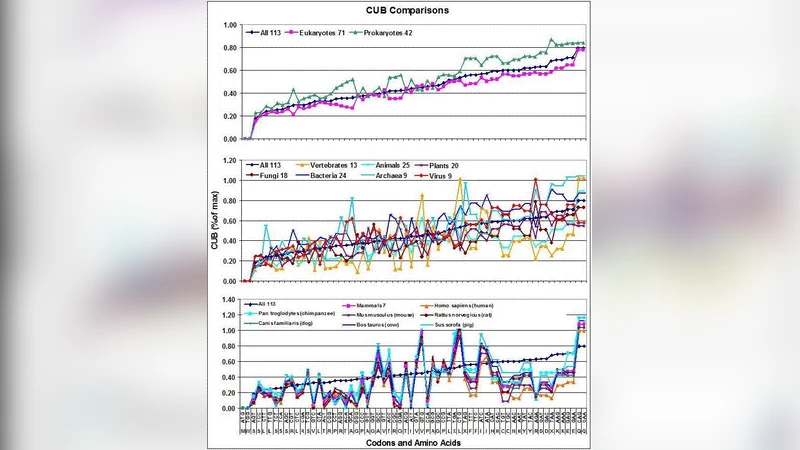
CUB is defined as the absolute deviation of observed codon frequencies from a theoretical uniform usage within each synonymous family. The maximal possible CUB is calculated as 2416.7 % (when only one synonymous codon per amino acid is used). Using this metric, the average CUB across the 113 species is 29.3 % ± 1.1 % (SEM). A clear trend emerges: prokaryotes (bacteria and archaea) display the highest bias, while eukaryotes, especially vertebrates and humans (CUB ≈ 18.9 %), show the lowest bias. This pattern is interpreted as a progressive reduction of codon bias with increasing genome and proteome complexity during evolution.
Statistical analyses reveal two robust correlations: (1) the number of synonymous codons per amino acid (n_i) positively correlates with the overall frequency of that amino acid in the proteome; (2) n_i also positively correlates with the magnitude of CUB for that amino acid. These relationships hold both in the aggregated pan‑genomic table (64 values) and in the species‑specific data (113 × 21 values).
A novel aspect of the study is the exploration of internal dynamics among codons. By sorting CUB values according to sign and magnitude, the author partitions codons and species into two complementary groups (Ac‑As/Bc‑Bs and Ac‑Bs/Bc‑As). Codons that are over‑represented in one group are under‑represented in the opposite group, indicating a pervasive inverse relationship that is consistent across all examined taxa.
Further, the paper examines nucleotide usage frequencies (NUF) at each codon position. Strong positive correlations are observed between the sum of complementary bases (A+T, G+C) in one position and the same sum in the other two positions. Most strikingly, the combined frequency of A and T in the first position predicts the frequency of A in the third (wobble) position, and analogous relationships exist for G and C. Leveraging these positional correlations, the author predicts the frequencies of the four possible wobble bases for all 64 codons. Prediction accuracy is assessed in four ways: (i) codon‑wise correlation of predicted versus observed frequencies (significant in 54 of 64 codons); (ii) species‑wise correlation (significant in all 113 species, p < 10⁻⁷ in most cases).
Methodologically, the study relies on Pearson correlation coefficients and linear regression, with randomised control datasets employed to confirm that observed correlations are not artefacts of sample size or distribution. However, the manuscript lacks multiple‑testing correction, effect‑size reporting, and direct comparison with established CUB metrics such as the Codon Adaptation Index (CAI) or Effective Number of Codons (ENC). The evolutionary interpretation of decreasing CUB is presented without phylogenetically independent contrasts, leaving open the possibility that observed patterns reflect taxonomic sampling rather than genuine evolutionary trajectories.
In summary, the paper provides an ambitious, data‑driven portrait of codon usage bias, highlighting (1) a measurable decline of bias with organismal complexity, (2) strong links between synonymous codon number, amino‑acid abundance, and bias magnitude, (3) pervasive inverse relationships among codons within genomes, and (4) the feasibility of predicting wobble‑base frequencies from first‑ and second‑position nucleotides. While the findings are intriguing and the pan‑genomic approach is valuable, the study would benefit from more rigorous statistical validation, integration with existing codon bias frameworks, and phylogenetically controlled analyses to substantiate its evolutionary claims.
Comments & Academic Discussion
Loading comments...
Leave a Comment