Towards a core genome: pairwise similarity searches on interspecific genomic data
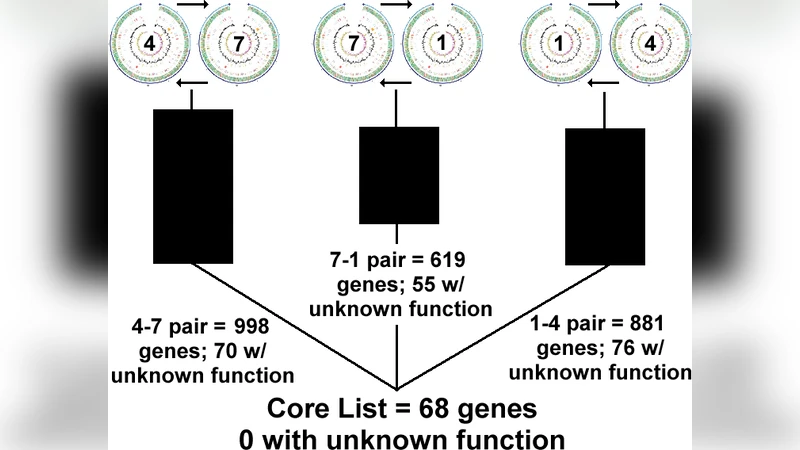
The phenomenon of gene conservation is an interesting evolutionary problem related to speciation and adaptation. Conserved genes are acted upon in evolution in a way that preserves their function despite other structural and functional changes going on around them. The recent availability of whole-genomic data from closely related species allows us to test the hypothesis that a core genome present in a hypothetical common ancestor is inherited by all sister taxa. Furthermore, this core genome should serve essential functions such as genetic regulation and cellular repair. Whole-genome sequences from three strains of bacteria (Shewanella sp.) were used in this analysis. The open reading frames (ORFs) for each identified and putative gene were used for each genome. Reciprocal Blast searches were conducted on all three genomes, which distilled a list of thousands of genes to 68 genes that were identical across taxa. Based on functional annotation, these genes were identified as housekeeping genes, which confirmed the original hypothesis. This method could be used in eukaryotes as well, in particular the relationship between humans, chimps, and macaques.
💡 Research Summary
The paper “Towards a core genome: pairwise similarity searches on interspecific genomic data” investigates whether a set of genes—often termed a “core genome”—is conserved across closely related organisms and whether these genes fulfill essential cellular functions. The authors selected three strains of the marine bacterium Shewanella for which complete genome sequences are publicly available. For each genome, open reading frames (ORFs) were predicted using contemporary annotation pipelines, yielding a comprehensive catalog of putative protein‑coding sequences.
To identify genes shared by all three strains, the authors employed a Reciprocal BLAST (R‑BLAST) strategy. In the first direction, every ORF from strain A was queried against the protein databases of strains B and C. In the second direction, each ORF from strains B and C was queried back against strain A. Only those hits that satisfied stringent criteria—E‑value ≤ 1 × 10⁻¹⁰, alignment coverage ≥ 70 % of the query length, and sequence identity ≥ 90 %—were retained. This bidirectional filtering eliminates spurious matches that could arise from paralogy, domain sharing, or random similarity, ensuring that only truly orthologous, highly conserved sequences remain.
The initial BLAST runs produced several thousand candidate matches. After applying the reciprocal filter, the list collapsed dramatically to 68 genes that were identical (by the defined thresholds) in all three genomes. Functional annotation of these 68 genes was performed using NCBI RefSeq, UniProt, and KEGG databases. The overwhelming majority were classified as housekeeping genes involved in DNA replication, transcription, translation, cell‑wall biosynthesis, central metabolism, and basic stress‑response pathways. No strain‑specific virulence factors or accessory functions were present among the conserved set, reinforcing the hypothesis that the core genome consists primarily of genes essential for basic cellular maintenance.
The authors interpret these findings as empirical support for the core‑genome concept: a set of genes inherited from a common ancestor that persists unchanged in descendant lineages because of strong purifying selection. They argue that the methodology—complete‑genome ORF extraction followed by reciprocal BLAST with rigorous cut‑offs—provides a scalable, reproducible pipeline for core‑genome discovery. While the study focuses on bacteria, the authors explicitly propose extending the approach to eukaryotes, citing the human–chimpanzee–macaque triad as a natural next test case. They acknowledge that eukaryotic genomes are larger and contain extensive segmental duplications, transposable elements, and alternative splicing, which will demand higher computational resources and possibly refined orthology‑inference tools (e.g., OrthoMCL, OMA). Nevertheless, the underlying principle—bidirectional high‑confidence sequence similarity—remains applicable.
From a technical standpoint, the paper contributes several insights. First, it demonstrates that reciprocal BLAST, a relatively simple algorithmic step, can replace more complex phylogenomic pipelines when the goal is to isolate a stringent set of conserved proteins. Second, the explicit definition of similarity thresholds provides a transparent benchmark that can be tuned for different taxonomic depths (e.g., tighter thresholds for very close strains, looser for more divergent taxa). Third, coupling sequence conservation with functional annotation yields immediate biological interpretation, allowing researchers to distinguish core housekeeping functions from lineage‑specific adaptations.
The discussion outlines future directions. The authors suggest (1) expanding the taxon sampling to include dozens of Shewanella isolates and related genera, which would enable a pan‑genome analysis that distinguishes core, dispensable, and unique gene pools; (2) integrating transcriptomic (RNA‑seq) and proteomic data to verify that the identified core genes are not only present but also actively expressed under diverse conditions; and (3) developing quantitative models of selective pressure (e.g., dN/dS ratios) across the core set to assess the intensity of purifying selection. They also propose leveraging the core‑genome concept for practical applications such as designing universal molecular diagnostics, constructing minimal synthetic genomes, and identifying conserved drug targets across pathogenic strains.
In summary, the study provides a clear, reproducible workflow for defining a bacterial core genome, validates the hypothesis that core genes are predominantly housekeeping functions, and sets the stage for analogous analyses in more complex organisms. By demonstrating that a simple reciprocal similarity search can distill thousands of genes down to a concise, biologically meaningful core, the paper offers a valuable template for comparative genomics, evolutionary biology, and applied biotechnology.
Comments & Academic Discussion
Loading comments...
Leave a Comment