An efficient strategy to characterize alleles and complex haplotypes using DNA-markers
We consider the problem of detecting and estimating the strength of association between a trait of interest and alleles or haplotypes in a small genomic region (e.g. a gene or a gene complex), when no direct information on that region is available bu…
Authors: Rodrigo Labouriau, Poul S{o}rensen, Helle R. Juul-Madsen
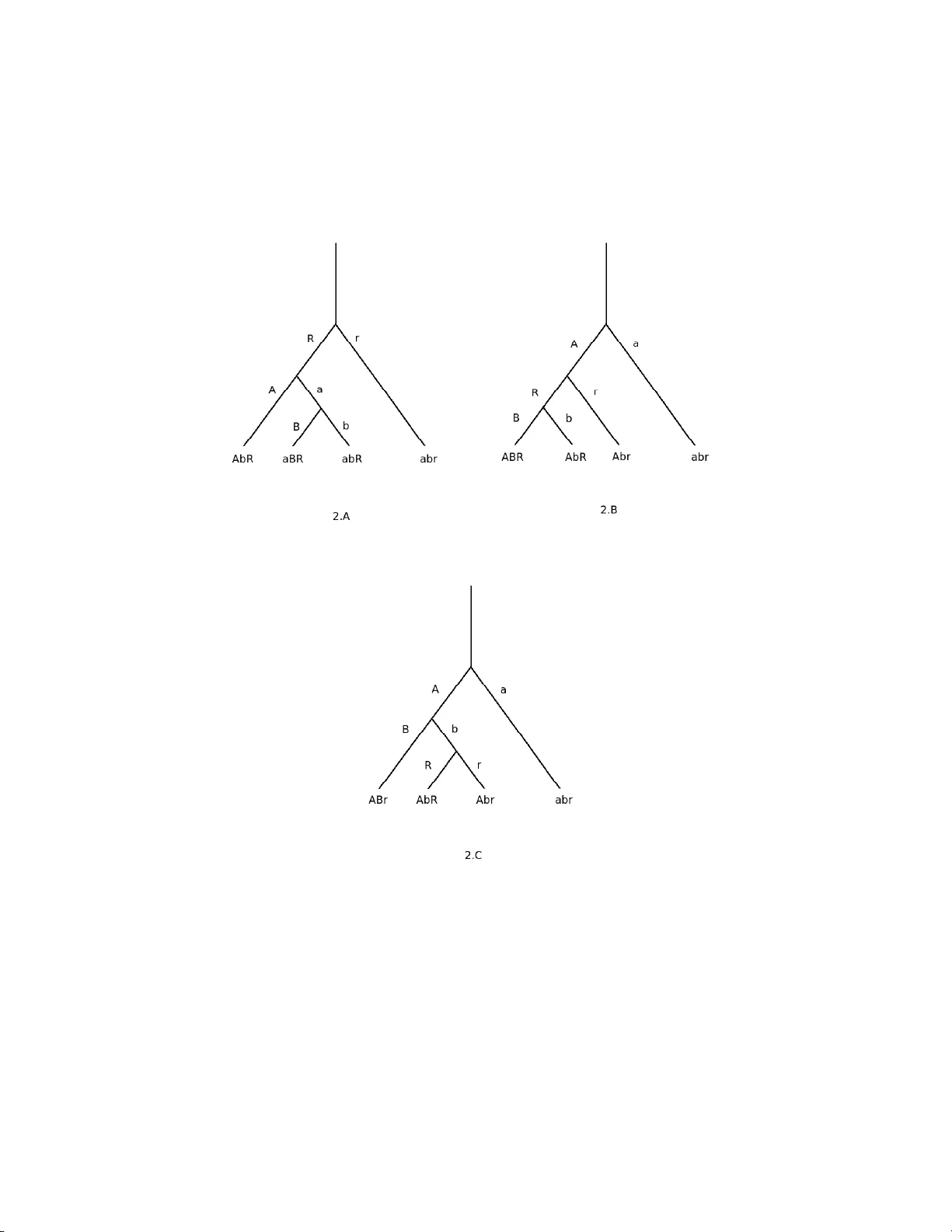
An efficien t strategy to c haracterize alleles and complex haplot yp es using DNA-mark ers Ro drigo Lab ouriau ∗ , P oul Sør ensen † , Hell e R. Juul-Madsen ‡ April 200 8 Key-w ords: Gener alize d Line ar Mo dels (GLIM), phylo genetic-tr e e, Akaike- information, Genetic-asso ciation, MHC Abstract W e consider the problem of detecting and estimating the s trength of asso ciation bet ween a trait of interest and alleles or haplotypes in a small genomic region ( e g a gene or a gene complex), when no direct information on that region is av aila ble but the v alues o f neig h b ouring DNA-markers are at hand. W e argue that the effects of the non-observ able haplotypes of the ge- nomic reg ions can and should be repres e nted b y factors representing disjoin t groups of mark er -alleles. A theor e tical ar gumen t base d on a hypothetical ph ylo genetic tr ee supp orts this genera l cla im. The techniques descr ibed allow to identify and to infer the n um b er of detectable haplotypes in the geno mic region that are asso ciated with a trait. The methods pr oposed use an exhaustive combinatorial s earc h coupled with the maximiza tion of a version of t he likelih o o d function penalized for the n umber o f parameters. This pro cedure can easily be implemented with stan- dard s tatistical metho ds for a mo derate n umber of ma rk er-a lleles. ∗ Research gr o up for B io informatics Genetics and Statistics, Department of Genetics and Biotechnology , F aculty of Agricultura l Sciences, Universit y of Aarhus. Corresp onding author: rodri go.labou riau@agrsci.dk † Research group for Population Genetics and E m bryology , Departmen t of Genetics a nd Biotechnology , F aculty of Agricultural Sciences , Universit y of Aarhus. ‡ Research g roup for Disease mechanisms, -markers and - prev ention, Department of Animal Health, W elfare a nd Nutrition, F aculty of Agricultural Sciences, Universit y o f Aarhus. 1 Efficien t characteriz ation of alleles and haplot yp es 2 1 In tro duct ion Often in genetic applications, and in special in imm une-genetics, in terest lies in detecting a nd quan tifying the asso ciation of a giv en genomic region to a trait. Here the genomic region migh t b e a gene or a gene-comple x. T yp- ically , no bio- molec ular or genomic information is directly a v ailable ab out this genomic region, but instead, a system of DNA-marke rs lo cated close to the region is used. An example of this situation is the study presen ted b y Sc hou et al , (2007, 2008) where the p ossible asso ciation of the suscept ibil- it y to sev eral parasites and the ma jor histo compatibilit y complex (MHC) in c hic k ens was inv estigated using the microsatellite LEI0258 as a mark er. An- other examp le in v olv es the use of a tig ht sys tem of SNP mark ers to asso ciate putativ e alleles in the MHC region and susceptibilit y to psoriasis in humans (Orru et al , 200 2 ). The association b etw een alleles or haploty p es in the genomic region o f in terest and the tra it is commonly c haracterized by a regression-lik e statis- tical mo del in whic h the trait enters as the dep enden t v ariable and factors represen ting the prese nce of the mark er-alleles are included among the ex- planatory v ariables. A common practice is to declare asso ciation b etw een the trait and the genomic region when at least one of the parameters represen ting the mark er-alleles is statistically significan t. In order to establish asso ciation b et w een traits and putativ e haploty p es in the genomic region of in terest it is required to use a represen tat io n of those haplot yp es in terms of mark er-alleles (the only genomic information av ail- able). This represen tation is crucial to prop erly c haracterize the asso ciation. W e argue that suc h a represen tation should b e constructed with groups of mark er-alleles instead of only individual marker alleles. Informally , our main p oin t is that when consid ering only groups consisted of single marker-alleles (as usually done when using a naiv e approach) one might fail to represen t alleles or haplotypes in the neigh b ouring genomic region. This leads to a sig- nifican t reduction of efficiency or ev en to the complete loss of the capacit y to detect certain asso ciations. Our approac h requires the use o f a more complex statistical inferenc e in v olving a searc h in a large n um b er of possibilities. W e sho w how ev er, that the statistical inference is feasible for a mo derate n um b er Efficien t characteriz ation of alleles and haplot yp es 3 of mark er-alleles (10- 15 allele-mark ers). The text is organized as follo ws. Section 2 con tains the basic setup, including a description of the genetic and molecular biological scenario, a form ulation of the statistical mo del in terms of a generalized linear model and some discussion on the prop er form of p erforming inference under those premises . A ph ylogenetic based argumen t justifying our prop osal is presen ted in the last part of section 2. The details of the implemen tation of the statis- tical inference are giv en in section 3 and one examples is discusse d in section 4. Some discussion is pro vided in section 5. 2 Setup 2.1 A genetic and molecular b iol ogical scenario W e a ssume that the data av ailable consist of observ ations on n diploid in- dividuals from a give n p opulation for whic h w e ha v e the information o n the v alues of a trait and a range of explanatory v ariables charac terizing the indi- viduals. The inte rest is in detecting and characteriz ing a p ossible asso ciation b et w een the trait of intere st and a lleles or haplotypes in a giv en genomic region suc h as a gene or a gene complex whic h are not directly observ able. W e will refer to these alleles or haplot yp es as the haplotyp e s in the genomic r e gion of in ter est . W e a ss ume additionally that data on D NA marke rs lo cated close to the genomic region is av ailable. These markers are assumed to b e tigh t link ed so that they can b e view ed as a single lo cus with sev eral p o ss ible a llele s ( e.g . a microsatellite mark er or a system of v ery close SNP mark ers), called the marker-al leles . The data av ailable can b e though t as comp osed of n tr iplets, ( y i , x i , m i ) , for i = 1 , . . . , n , where i indexes the n individuals, y i is the v alue of the trait, x i = ( x 1 i , . . . , x k i ) is a v ector of auxiliar explanatory v ariables and m i = ( m 1 i , . . . , m H i ) is a v ector represen ting the v alues of H allele-mark ers o bserv ed for the i t h indi- vidual. Efficien t characteriz ation of alleles and haplot yp es 4 2.2 The basic statistical mo del W e in tro duce b elo w a suitable generalized linear mo del (GLIM) that will serv e as a framework to exp ose our metho d. It is straigh tforw ard to extend the tec hniques presen ted to other regression-lik e statistical mo dels. The generalize d linear mo del describing the data is specified by c ho osing a distribution f or the trait among the fa mily of the expo nen tial disp ersi on mo dels ( Jø rg ens en et a l , 1996) (t ypically , but not necessarily , a normal dis- tribution) and sp ecifying a relationship b et w een the exp ecte d v alue of t he trait and the explanatory v ariables ( x and m ). Here w e assume that there is a smooth monotone function g , called the link f unc tion , and the parameters β = ( β 1 , . . . , β k ) T and α = ( α 1 , . . . , α h ) T suc h that g [ E ( y i | x i , m i )] = x i β + α 1 I m 1 i + . . . + α h I m h i , (1) where I m j i is an indicator v ariable taking the v alue 1 if t he i t h individual carries the j t h mark er-allele a nd 0 otherwise. W e assu me, for simplicit y , that all the alleles act as completely dominan t. That is, the effect of an allele in homozygous individuals carrying tw o copies of the allele is equal to the effect of the allele in heterozy gous individ uals carrying one cop y of the allele. This assumption can easily b e mo dified to include o t her genetic mec hanisms b y mo difying the definition of the factors repre sen ting the mark er-alleles ( e g b y defining factors with more than t w o lev els for represen ting partial dominance). Using standard tech niques of generalize d linear mo dels it is p ossi ble to mak e inference on the parameters α and β . Here our in terest lies in the pa- rameter α represen ting the effects of the marke r-a llele s, while β is considered as an auxiliary nuis ance parameter. The asso ciation b et we en the genomic region of in terest and the trait is often v erified by conside ring a test of h yp othesis give n b y H 0 : α = 0 × H a : α 6 = 0 . Since 0 represen ts a v ector with all comp onen ts equal to zero, the nu ll h y- p othesis H 0 ab o v e is say ing that all the componen ts of the vec tor α are equal to zero while the alternativ e h yp othesis H a states that at le ast o n e of Efficien t characteriz ation of alleles and haplot yp es 5 the mark er alleles has a non-v a nishing effect. This test can b e easily carried out b y comparing a mo del defined b y (1) to a mo del defined b y g [ E ( y i | x i , m i )] = x i β , (2) using a lik eliho o d ratio type test. Rejection of the nu ll h yp othesis indicates asso ciation of the genomic region in study to the trait of in terest. Although this simple join t test detects association, it do es not help to iden tify the asso ciated haplot yp es in the g enomi c region of in terest. A naiv e pro ced ure is to iden t if y alleles or haploty p es in the genomic region b y lo oking fo r the mark er-alleles with statistically signifi cant effects. W e claim that this can b e misleading. It migh t happ en that some of the alleles or haplot yp es in t he genomic region are in close linkage disequilibrium ( i.e. are tigh t link ed to) with more than one mark er-allele in suc h a a w a y that in some individuals the first mark er-allele o ccur while the second do not o ccur (and vice-v erse). A ph ylogenetic-based argumen t presen ted b elo w sho ws that this scenario can and indeed of ten o ccurs . Under this situation, the tests for the effect of each single factor represen ting each of t he mark er-alleles would not ha v e biological meaning and w ould imply in a loss of p ow er due to a misclassi fication of individ uals. Therefore, the inference o n haplot yp es in the genomic region of in terest should b e p erformed using sets of mark er-alleles instead of only individual mark er-alleles. The precise formal statemen t of this idea is giv en b elo w. Let G 1 , G 2 , . . . , G H b e pairwise disjoint non-empt y subsets of the set of marker alleles { m 1 , . . . , m h } (with H ≤ h ). Define the mo del giv en by g [ E ( y i | x i , m i )] = x i β + α 1 I G 1 i + . . . + α H I G H i , (3) where I G j i is a v ariable taking v a lue 1 if the i t h individual carries at least one allele-mark er b elonging to the subset G j (for j = 1, . . . , H ). Cle arly , the simple mo del give n b y (1) is con tained in the class of mo dels in the form give n b y (3), since the disjoin t subsets G 1 , G 2 , . . . , G H can b e a ll constituted of a single elemen t. Ho w ev er, this class of mo dels con tains man y other mo dels (an y p ossible combination of non-empt y disjoin t subsets of G ), whic h op ens the p ossibilit y of finding a mo del of this ty p e that suitably describ es the genetic phenomena in play . W e discuss in section 3 a strategy for searc hing for the b est represen ta tio n among the man y p ossibilities. Efficien t characteriz ation of alleles and haplot yp es 6 2.3 A ph ylogenetic-based argumen t A nu mber of sp ecial structures naturally app ear during the ev o lution pro cess of a population. As a conseq uence, the information that DNA-mark ers carry on neigh b our lo ci is distributed according to some c haracteristic patterns. In this section w e illustrate this general claim using a simple ph ylogenetic- lik e construction based on dic hotomous branc hing trees. W e will sho w how some motifs o f asso ciation inv olving mark ers and alleles in the genomic region o ccur naturally . This will then b e used to argue in f av or of using a prop er represen tat io n of the effect of DNA-markers a nd to base the inference using statistical mo dels defined with groups of mark er-a lleles as in (3). Consider a lo cus R in the genomic region of in terest a nd t w o observ able mark ers A and B in a neigh b ourho o d of R . Supp ose, for exposition simplicit y that R , A a nd B are di-allelic with pairs of alleles ( R, r ) , ( A, a ) and ( B, b ) respectiv ely . Assume , moreo v er, that r ecombin ation b et w een these lo ci can b e neglected due t o a strong linkage disequilibrium a r o und the region of in- terest. W e can think o f eac h of those a llele s as the result of a single ev en t o ccurred at some p oin t in the evolu tionary history of the population in pla y ( e.g. a single nucle otide mutation or the duplication of a small genomic re- gion). The sequence of eve nts that generated these alleles can b e represen ted b y a tree with three dic hotomous branchi ng, eac h branchin g corresp onding to the ev en t that generated one of the alleles. W e use the con v en tion that the alleles represen ted b y capital letters are the results of ev en ts, while the alleles represen ted b y small letters are the reference alleles, or wild types, correspo ndin g to the states of the lo ci b efore the ev en ts. A mark er-allele A c arries inform a tion ab out the lo cus R when the kno wl- edge of the o ccurrence of A determines the allelic form of R . If the allele A can o ccur together with the allele R and the allele r , then A is said to b e neu- tr al with resp ect to the lo cus R . F or instance, if the branc hing that formed the lo cus R o ccurred b efore the branc hing of the lo cus A and the branc hing of A o ccurred in the branch of the tree containi ng the allele R (see F ig ure 1A), then the o ccurren ce of the allele A in the lo cus A implies that the lo cus R carries the allele R . Moreov er, in that circumstance s t he o ccurre nce of the allele a do es not imply neither that R carries the allele R nor r . Therefore the a llel e A carries information on the lo cus R and the allele a is neutral Efficien t characteriz ation of alleles and haplot yp es 7 with resp ec t to R . Figure 2A illustrates a scenario where the branc hing of the lo cus R o c- curred first, whic h w as follo w ed b y the branc hing of the lo cus A and then the branc hing of the lo cus B . Moreo ve r, the branc hing of the lo cus A o ccurred in the branc h of the tree con taining the allele R while the branching of the lo cus B o ccurred in the branc h of the tree con taining the mark er-allele a . Under these circumstances there are only four p ossible haplotypes: AbR , a BR , abR , abr . The allele A o nly o ccu rs together with the allele R , and it carries infor- mation about the lo cus R . Analogously , the allele B carries also information on the lo cus R . Since the alleles a and b might o ccur together with b oth the allele R a nd the allele r , then b oth a and b are neutral with resp ect to the lo cus R . W e can still draw further conclusions ab out the distribution of the information o n the lo cus R . If w e w ant to use a rule for detecting the pres- ence of the allele R based on of the o ccurrence of mark er-alleles, then the rule ’ R o ccurs when A or B o ccurs’ w ould detect t w o out of the three o ccurren ces of the allele R . A rule based only on the o ccurrence of the mark er-allele A w ould only detect o ne o ut of the three p ossible o ccurrences of the allele R and therefore w ould b e less efficien t in detecting R than the rule using the alleles A and B together. The alleles a and b are b oth neutral and therefore the o ccurrenc e of the allele R in the haplotype a bR cannot b e detected using the information con tained in the mark er-alleles. W e conclude that under the scenario describ ed b y Figure 2A, one can only detect the o ccurrenc e o f the allele R using a rule based on the mark er-alleles in t wo out of the three p os- sible haplot yp es con taining R . This maxim um p ossible information recov ery is attained only by the rule ’ R o ccurs when A or B o ccurs’. A different scenario is described in Figure 2B where the branching in the lo cus A o ccurred first, follo we d by the branching in the lo cus R , in the branc h con taining the allele A , and then the branc hing in the lo cus B in the branch of R con taining the allele R . In this case the four po ss ible haplot yp es are: ABR , AbR , Abr and a br . Therefore the mark er-allele B carries information on R and the mark er-allele A is neutral. The alleles a and b necessarily o ccur together a nd b oth carry information on R (but the same information). Here there are tw o cases in whic h the genot yp e of the lo cus R is determined b y the genot yp es of the marke r- allele s: ’o ccurrence of B ’ and ’o ccurrence of a and Efficien t characteriz ation of alleles and haplot yp es 8 b ’ implying the o ccurrence of R or r resp ectiv ely . Note that the last ev ent ’o ccurrenc e of a and b ’ is equiv a len t to t he eve nt ’not o ccurrence of A or B ’. Under this scenario there are o nly t w o rules based on the mark er-a lleles genot yp es that determine the genoty p e at the lo cus R , bo th can b e expressed as the effect of a comb ination of the o ccurren ce of the mark er-alleles A and B . The first rule (’o ccurren ce of B implies o ccurren ce of R ’) can a lso b e expressed as the effect of a single allele-mark er as in the tr a ditional inference metho d, but the second rule (’not o ccurrence of A or B implies the o ccurrence of r ’) requires the use of groups of mark er-alleles as in the models described in (3) to be prop erly r epresen ted in a statistic al mo del. Here stic king only to rules based on single mark ers w ould represen t a loss of half o f the p ossibilities for determining the allele at the lo cus R , that is a loss o f half of t he information on the genot yp e of the lo cus R that could b e recov ered with the know ledge of the mark er-alleles. Figure 3 displa ys the branchin g tree of a more complex scenario comp osed with the lo cus R in the genomic region of in terest and four mark er-lo ci A , B , C , and D , with alleles A , a , B , b , C , c , D and d resp ectiv ely (w e apply the same notational con v ention as b efore). The follo wing six haplotypes are formed: AbcdR , aBCdR , aBcdR , abcDR , abcdR and abcdr . Therefore, the rule ’if A or B or C or D o ccur then R o ccurs ’ detects f o ur out of the fiv e haplot yp es con taining the allele R . Moreov er, under the curren t scenario, this is the b est p ossible rule that could b e constructe d with the information on the mark er-alleles that w e hav e at hand. Although the allele C carries information on the lo cus R , this informatio n is redundan t si nce C o ccurs alw a ys together with B . W e can then remov e the o ccurrenc e of the allele C from the rule and still detect the same cases where the allele R o ccurs when using the rule including the marker-allele s of the four mark er lo ci. The discussion ab o v e sho ws that in sev eral situations the use of a naiv e represen tat io n of the effects of the mark er-alleles is inefficien t and that fully efficiency is obtained when using the approach in v olving the represen tation of groups of allele-mark ers. This phenomenon is not restricted to the few scenarios presen ted here. W e claim, without giving a formal pro of, that ev ery time that there is a branc hing after the branchin g that generated the allele in the lo cus R the new mark er-allele formed will carry information on the lo cus Efficien t characteriz ation of alleles and haplot yp es 9 R . M oreov er, if the branc hing o ccurs in the branc h that con tains the wild t yp e allele of the last branchin g of a mark er lo cus, then the new mark er allele formed will add new informatio n on the lo cus R that is not con tained in the mark er-alleles formed b efore. This prog ressiv e gain of information obtained while the new mark er-alleles are being formed is only fully realized if w e use a rule of the ty p e ’if A or B or C or ... o ccurs, then R o ccurs’. Figure 1: T wo dic hotomic phylogenetic trees inv olving a lo cus R in the geno mic regio n of interest with the alleles R (v a rian t allele) and r (wild type allele) a nd one mar k er lo ci A with alleles A and a . The haplotypes formed at each ending branch are displayed at the bo ttom of the tree. Efficien t characteriz ation of alleles and haplot yp es 10 Figure 2: Three dic hotomic ph ylo genetic trees involving a lo cus R in the g enomic region of interest with the alleles R (v ariant allele) and r (wild t yp e allele) and t wo marker lo ci A and B with alleles A , a , B , b respec tively . The haplot yp es for med at each ending branch are displayed at the bo ttom of the tree. Efficien t characteriz ation of alleles and haplot yp es 11 Figure 3: A complex dichotomic phylogenetic tre e, inv olving a lo cus R in the genomic region of in teres t with the allele s R (v ariant allele) and r (wild type allele) and fo ur marker lo ci A , B , C and D . The haplotypes formed at ea c h ending bra nc h a re display ed at the bo ttom of the tree. Efficien t characteriz ation of alleles and haplot yp es 12 3 Inference w ith a mo derate n um b er of mark er- alleles 3.1 Exhaustiv e searc h strategy The strategy we prop ose for c haracterizing the asso ciation b et we en a genomic region and a trait consists in searc hing exhaustiv ely all the p ossible groupings formed with sets of mark er-alleles and then c ho ose t he b est candidate among the many p ossibilities . Here the b est candidate is one that represen ts all the haplot yp es of the genomic region of in terest that are associated with the trait and that is not redundan t. W e define a gr ouping of the marker-al leles as a collection of non-empt y disjoint subsets of the set of all marker-allele s. The subs ets of a grouping are called marker-al leles gr oups (MA G). The idea is to use MA Gs to r epresen t haplotypes in the genomic region of interes t that might b e asso ciated with the tr a it. F or eac h p ossible grouping of the mark er-alleles one statistical mo del of the type described b y (3) containing factors represen ting eac h MAG o f this grouping is fit. The grouping that generates the mo del with the b est fit, according t o a criterion to b e defined b elo w, is c hosen to represen t the asso ciation b et w een the genomic region of in terest and the trait. The grouping will b e c hosen in suc h a w ay that it will not contain redundancy , so eac h MA G will represen t one haplot yp e in the region of in terest. The n umber o f MA Gs in this grouping will b e the n um b er of detectable haplot yp es asso ciated with the trait. The magnitude of the effect of eac h MAG will b e then the comp onen t of the mag nitud e o f the haplot yp e that is detec table through the mark er-alleles (whic h is smaller or equal to the magnitude of the effect o f the unkno wn haplotype). This pro cedure is only feasible if the n umber of mark er-alleles is not v ery large (w e were able to analyse a data with 9 allele marke rs in few minu tes in an ordinary p ersonal computer). It is conv enien t to mak e the exhaustiv e en umeration o f a ll p ossible group- ings of the marke r- allele s in the follo wing wa y . Consider the class P of all subsets of the set of mark er-alleles G = { m 1 , . . . , m h } . F o r mally , P is the class of parts of G . W e asso ciate one statistical mo del to eac h elemen t c of P b y defining the mo del of the type defined by (3) that incorp orate fa ctors Efficien t characteriz ation of alleles and haplot yp es 13 represen ting the groups of mark er-alleles prese nt in c . F or j = 1, . . . , h let P j b e the class of all the subsets of G containing exactly j disjoint non-empt y sub-sets. Clearly P is the disjoin t union P = P 1 ∪ . . . ∪ P h . (4) Therefore we can searc h for the b est mo dels by pro ceeding in tw o steps: F irst w e find the b est mo del for eac h P j ( j = 1, . . . , h ) and then w e find the b est mo del among the candidates found in the first step. The selec tion of the b est mo del related to a giv en P j ( j = 1, . . . , h ) is done b y c ho osing the mo del with the largest v alue of the lik eliho o d ( o r equiv alen tly the log-lik eliho o d) function. In this w ay the set of v alues of the lik eliho ods of the c hosen candidate fo r eac h P j is a profile set and pla ys a rule analogo us to the rule of a profile lik eliho o d curv e for the num b er of marker-allele s. Denote the mo del that attains the maxim um of the lik eliho o d for a giv en P j b y M j and the v alue log-lik eliho o d f unction of M j at t he maxim um b y ˆ l j (for j = 1, . . . , h ). W e refer to these quan tities as the pr o fi le mo del and the pr ofile lo g-likeliho o d of order j . 3.2 Determinati on of the n um b er of detectable asso ci- ated haplot yp es The next step in the pro ced ure of inference is to c ho ose the class P j ( j = 1, . . . , h ) that yields the b est statistic al mo del. If w e assume that the haplo- t yp es in the genomic region of inte rest are represen table in terms o f subsets of G , then c ho osing the class P j that pro duces the b est statistical mo del is equiv alen t to infer the n um b er of detectable haplotypes in the genomic region of in terest. The profile lo g -lik eliho o d nev er decrease s when the n umber of haplot yp es assumed in the mo del increase, i.e. ˆ l 1 ≤ . . . ≤ ˆ l h , (5) since a mo del M i ( i < h ) can b e expressed as a sub-mo del of a mo del with i + 1 MA Gs in whic h a pa ir of MA Gs presen t the same effect. As a conseque nce, it is not reasonable to estimate the num b er of haploty p es in Efficien t characteriz ation of alleles and haplot yp es 14 the region o f inte rest by c ho osing the P j with larger lo g -lik eliho o d. W e argue next that maximizing the negativ e Akaik e information (or a v ariant o f it) is a reasonable pro cedure for inferring the num b er of haplot yp es in the g enomi c region of in terest. The inequalit y (5) do es not extract all t he information a v ailable on the de- v elopmen t of the profile lik eliho o d curv e as the num b er of putativ e haplot yp es of the genomic region of intere st increases. Indeed, the profile log-lik eliho o d curv e is expected to increase significan tly with the n um b er of putativ e haplo- t yp es until the n um b er o f detectable haplotypes is reache d. After that p oin t the profile log-lik eliho o d curv e is supp osed to remain appro ximately constan t. T o see that, consider the situation where there are J asso ciated haplot yp es in the genomic region. If j < J , then the mo del M j fails to represen t at least one haplot yp e and then the profile log- lik eliho o d should increase in a statis- tically significan t w a y with the addition o f the p o ss ibility to represen t one more haplot yp e. O nce a ttained the num b er of haplotypes the gain obtained b y increasing the capacit y of the mo del to represen t one more haplot yp e v an- ishes and only marginal ga ins in the profile log- lik eliho o d are exp ected (see figure 4). W e assume implicitly here that there are no significan t mixtures in the da ta and that the mo del is not missing an y imp ortan t explanatory v ariable (see Figure 4) The informal argumen t ab o v e suggests w e can infer the num b er of detectable haplot yp es in the genomic region b y searc hing for the p oin t at whic h the profile lo g -lik eliho o d curv e remains (approx imately) constan t. One w ay to do that is to subtract a suitable quan tit y from the profile log- likeli ho o d. By doing that, the new adjusted profile log-like liho o d w ould decrease approx imately linearly in the region where the original profile log-lik eliho o d w as constan t ( i.e. when the a ss umed n um b er o f haploty p es is larger than the n um b er of detectable haploty p es in the genomic region of in terest). If the subtracted quan tit y is not to o large, the adjusted profile log-lik eliho o d curv e will still increase in the region where the original pro- file log-lik eliho o d is increasing significan tly (b efore attaining the num b er o f detectable haploty p es). The so called Akaik e information criterion (Akaik e, 1974, Burnham and Anderson, 2002) explores this idea and is equiv alen t to subtract the n um b er o f parameters in the mo del from the log-lik eliho o d. In fact the Akaik e information A IC is defined as min us t wice the difference of Efficien t characteriz ation of alleles and haplot yp es 15 the log-lik eliho o d and the n um b er of parameters, more precisely , AIC = − 2 log ( ˆ L ) − k , where k is the num b er of parameters in the statistical mo del, and ˆ L is the maximized v alue of the lik eliho o d function for the estimated mo del. Mini- mizing the Akaik e information is equiv alen t to maximizing the log-lik eliho o d adjusted by subtracting the n um b er of parameters in the mo del. This appar- en tly arbitrary c hoice for the quan tit y subtracted from the log-like liho o d can b e j us tified as b eing equiv alen t to subtract f r o m a lik eliho o d ratio statistic its (asymptotic) exp ecte d v alue. Alternativ ely , one migh t subtract 3. 8 5k from the profile log-lik eliho o d whic h w ould b e equiv alent to p erform a like liho o d ratio test for incorp orating the represen tation of an additional haplot yp e to the curren t mo del when w orking at a significance lev el of 5%. Summing up, the pro cedure w e prop ose is to maximiz e the log - lik eliho o d in eac h class P i (whic h is equiv alen t to minimize the AIC in this class of mo dels since all the mo dels in P i has b y construction the same n um b er of parameters), for i = 1, . . . , h , and then choose the mo del with the smaller (profile) AIC (or equiv alen tly , with the larger adjusted profile log-lik eliho o d). 4 Example: C hic k en su sceptibilit y to helmin ths The asso ciation b et w een t he susceptibilit y to the helmin th A sc aridia g al li in c hic k ens and the ma jor histocompatibility (gene) complex (MHC) was in- v estigated in tw o rece nt studies (Sc hou et al , 2007, 2008). These studies used the microsatellite LEI0258 (F ulton et al., 20 0 6), whic h is lo cated in a non-co ding region b et w een t w o con tiguous regions of the MHC gene complex (the B-F/B-L and the BG lo ci), to obtain eigh t p olymorphic mark er-alleles here denoted 195bp, 207bp, 219bp, 25 1 bp, 264bp, 276bp, 2 97bp a nd 324bp. Since recom bination within the c hic k en MHC is very rare (Plac hy et al., 1 9 92; Miller et al., 2004 ), the alleles of the microsatellite LEI0258 are exp ec ted to b e in tigh t linkage disequilibrium with haploty p es formed by alleles at the Efficien t characteriz ation of alleles and haplot yp es 16 BF/BL and the BG lo ci ( i.e. the MHC haplot yp es). Moreo ver, we can dis- card the p ossibilit y of a direct effect of the LEI0258 alleles since this mark er is lo cated in a non-co ding region (as any microsatellite marke r). Therefore it is reasonable to apply the tec hniques described a bov e using the alleles of the microsatellite LEI0258 as marke r- a llele s in the set-up describ ed ab ov e. In the first study ( Schou et al , 2007) the in tensit y of infection with A. gal li w a s determined for birds of tw o chic k en breeds b y coun ting the n um b er of this w orms found in the in testine of each of the birds examined. The coun ts w ere categorized as, zero, low (up to 3 coun ts), in termediate (4 to 10 coun ts) and high (more than 10 coun ts). The cut-off p oin ts u sed for defining the categories a bov e w ere c hosen in suc h a w a y that the losses of the Külbac k-Leib er information about the coun ts due to a dis cretization w ere minimized. The asso ciation b et w een the in tensit y of infection and the MHC haplot yp es w as studie d b y applying a baseline-category logits mo del for m ultinomial distributed data (Agresti, 199 0 ) using the zero-category as a reference. Inference in these mo dels can b e p erformed by fitting three logistic mo dels constructe d with a common reference category (Ag r esti, 1990) whic h can b e done b y using standard generalized linear mo dels (with a binomial distribution and a logistic link). The standard method was used in this study and an association w as declared if the effect of a mark er-allele, in the presenc e of the other mark er-alleles, w as statistically significan t. Using this pro cedure it w as found that the o ccurrence of the mark er-allele 276bp w as asso ciated with an increased resistance. No further asso ciations w ere found with the standard pro cedure. A second study (Sc hou e t al , 2008) w as indep enden tly p erformed with differen t birds of the same t wo c hick en breeds. In this study the birds w ere ino culated with A. gal li under controlle d experimen tal conditions and the fecal excretion of A. gal li eggs w as monitored. Eac h animal w as classified based on the coun ts of eggs a s presen ting zero, lo w, in termediate or high infection lev el. A baseline-category logits model w as applied but, differen tly from the first study , using the strategy of searc hing for mark er-allele gr o ups (MA G). Three mark er-allele groups, denoted MA G- 1 , MA G- 2 a nd MA G- 3 , w ere iden tified and found to be associated with the inte nsit y of infec tion with A. gal li . Figure 4 displa ys the profile log-like liho o d a nd profile Akaik e Efficien t characteriz ation of alleles and haplot yp es 17 information as a function of the num b er of MA Gs assumed to b e asso ciated to the infection lev el. A joint lik eliho o d r a tio test indicated a statistically significan t effect of these three MA Gs on the intens ity of infection (p=0.0013). MA G-1 w as formed b y the LEI025 8 alleles 297bp and 32 4 bp; MA G-2 w as comp osed of the alleles 195 bp, 207bp, 219bp and 264bp; and MA G-3 only con tained the allele 276bp. Detailed analyse s rev ealed that a nimals carrying MA G-1 or MA G-3 presen ted larger resistance to A. gal li , while MAG-2 w as asso ciated with augmen ted susceptibilit y (Sc hou et al , 2008 ). An a p o steriori analysis of the data of the first study using the strategy of searc hing for mark er-allele groups yielded the same significan t mark er- allele groups with MA G-1 and MA G- 3 asso ciated to resistance and MA G- 2 asso ciated to susceptibilit y to A. g al li (rep orted in Sc hou et al , 2008). Note that when applying the standard strategy only the effect of the marke r- a llele 276bp w as fo und significan t, whic h is equiv alen t of detecting the mark er-allele group MA G-3 (comp osed only by the allele 276bp). None of the mark er- alleles comp osing the marke r-a llel e gr o up MA G- 1 and MAG-2 ( i.e. 297bp, 324bp, 1 9 5bp, 207bp, 219bp a nd 264bp) presen ted an individual significan t effect, whic h illustrates the loss of p o w er to detect asso ciation when applying the standard mo delling strategy . Efficien t characteriz ation of alleles and haplot yp es 18 1 2 3 4 5 −210 −205 −200 −195 −190 Number of marker−allele groups Log−likelihood Profile log−likelihood Negative profile AIC Negative alternative AIC Figure 4: Pr ofile log-likelihoo d, negative profile Akaike information (da s hed and dotted line) and negative profile alternative Akaike infor mation (dotted line) as a function of the assumed num b er of marker-allele groups for the data o f the second study on s usceptibilit y to A. gal li (Schou et al , 200 8). The das hed hor izon tal line r epresen ts the lo g-lik eliho o d of a mo del containing the maximum num b er of marker-allele groups ( i.e. 8 ) ev aluated at its ma xim um, that is, an uppe r b o und for the v alues of the profile log -lik eliho od. The alternative Akaike informa tio n was obtained by subtra ctin g 3.85k for each succ e s siv e increase in the num b er of ma r k er-a llele groups, corr e s ponding to test the incorp oration of one mor e marker-allele group at a 5% level of significance. The v alue 10 was added all the v a lues of the negative alternative profile Akaike infor mation for gr aphical conv enience. The negative profile Akaike information and the alternative negative profile Akaike information presented bo th a ma xim um at 3 suggesting the presence of 3 detectable mar k er-a llele groups. Efficien t characteriz ation of alleles and haplot yp es 19 5 Discuss ion W e presen ted a strategy for p erforming statistical inference that allo ws to represen t t he o ccurrence of non-observ able alleles or haplot yp es in a genomic region of intere st in terms of a range o f observ a ble mark er-alleles at highly link ed lo ci. The kern el idea presen ted here is that the natural unit to build statistical mo dels under this conte xt are not the mark er-alleles but groups of marker-allele s. As argued due to the w ay haplotypes in a genomic r e- gion of relative ly small size (suc h that allo w us to ignore recom bination) are formed during the ev olution of a population, certain haplot yp es formed with mark er-alleles will o ccur naturally in tight linkage diseq uilibrium with (non- observ able) haplotypes in the genomic region. T he w ay these mark er-a llele haplot yp es are constituted imply that detection rules based on indication functions of groups of mark er-alleles are optimal in the sense that they allow to extract the maxim um p ossible amoun t of information that is con t a ined in the mark er-alleles. Naiv e represen tations constructe d exclusiv ely with groups of allele-mark ers with only one elemen t are b ounded to use inefficien tly the information (if not destro y it completely), a s illustrated in an example with real data. The tec hniques presen ted here in v olve fitting man y mo dels and selecting a b est candidate among the (ve ry) man y p ossibilities, fo llowin g a sequenc e of mo dels with increasing num b er of assumed mark er-allele groups. This or- der is to ugh to facilitate the inferenc e o f the n um b er of mark er-allele groups with de te ctable effect o n the trait of inte rest. Ho w ev er, this for c e brut ex- haustiv e searc h is not feasible for a large num b er of mark er-alleles, since the n um b er o f possibilities to b e c hec ke d increases v ery rapidly with the n umber of mark er-alleles. An alternativ e is to use Mon te Carlo base d a lgorithms for maximization in discrete parameter space a s sim ulated annealing or t he genetic algorithm. W e sho w ed that the classical criterion of maximizing the log-lik eliho o d cannot b e used to estimate the num b er o f detectable mark er-alleles groups, since the lik eliho o d function cannot decrease with the n um b er of MA Gs. A w a y aro und that is to use the Akaik e information criterion whic h p enalize s the log -lik eliho o d of models with to o many (unnecessary) parameters. Other Efficien t characteriz ation of alleles and haplot yp es 20 forms o f p enalized lik eliho o d could b e applied, as for instance the a lternativ e information criterion prop osed in the text, whic h is equiv alen t to p erform a lik eliho o d ratio test for incorp orating one extra MAG in the mo del (at a 5% lev el o f significance). T he c hoice of the p enalized lik eliho o d method to b e used dep end on the t yp e of basic statistical mo del used. W e used generalized linear mo dels to explain our ideas, but w e stress that other mo dels could ha d b een used without essen tially c hanging the pro cedure expo se d here. Indeed, the example presen ted uses in fact a sligh t extension of generalized linear mo dels. Probably one of the most relev an t extensions w ould b e the incorpo r a tion of random comp o nen ts allo wing to r epresen t po p- ulation structure, co-ancestry a nd p olygenic effects. Another p ossibilit y to b e explored is the incorp oration of information on a ncestors genot yp es a nd other mec hanisms of inheritance. In conclusion, the tec hniques pres en ted here ar e flexible and relativ ely easy to implemen t using classical statistical mo dels and standard soft w are. A c kno wledge men ts: This work o rig inated during the statistical analysis of the data collected by T orb en Wilde Sc hou. W e thank him f o r kindly allowing us t o presen t part of his data as an example. References [1] Akaik e, H., (1974). New Lo ok at Statistical-Mo del Iden tification. IEEE T r ansactions on A utomatic Contr ol 19 (6), 716-7 23. [2] Burnham, K. P ., and Anderson, D.R. (2002). Mo del Sele ction and Mul- timo de l Infer enc e : A Pr actic al-The or etic A ppr o a ch , 2nd ed. Springer- V erlag. ISBN 0- 387-95364-7. [3] F ulton, J.E., Juul-Madsen, H.R., Ash w ell, C.M., McCarron, A.M., Arth ur, J.A., O’Sulliv an, N.P . and T aylor, R.L., Jr.(2006). Molecular genot yp e ide ntific atio n of the Gallus gallus ma jor his to compatibilit y complex. Immuno genetics 58 (5-6), 407-421 . Efficien t characteriz ation of alleles and haplot yp es 21 [4] Jørgensen, B., Lab ouriau, R. and Lundb y e-Christensen, S. (19 96). Lin- ear gro wth curv e analysis based on expo nen tial disp ersion mo dels . J.R. Statist. So c. B 58 , No. 3, 573 - 592. [5] Miller, M.M., Bacon, L.D., Hala, K., Hun t, H.D., Ew ald, S.J., Kaufman, J., Zo orob, R. and Briles, W.E.(2004) Nomenclature for the c hic k en ma jor histocompatibilit y (B and Y) complex. Immuno ge n etics 56 (4 ) , 261-279. [6] Orru S., E. Giuressi, M. Casula, A. Loizedda, R. Murru, M. Mulargia, M.V. Masala, D. Cerimele, M. Zucca, N. Aste, P . Biggio, C. Carcassi, L. Con tu (2002). Psoriasis is associated with a SNP haplot yp e of the corneo desm osin gene (CDSN). Tissue A ntigen s 60 (4), 292-29 8. doi:10.1034/j.1399 -0039.2002.600403 .x [7] Plac h y , J., Pink, J.R.L. and Hala, K. (1992). Biology o f the Chic k en Mhc (B-Complex). Crit. R ev. Im m unol. 12 (1 - 2), 47- 79. [8] Sc hou, T.L.H, P ermin, A., Juul-Madse n H.R., Sørensen P ., Lab ouriau R., Nguy e T.L.H, Fink M. and Pham S.L. (20 0 7). Gastroin testinal helmin ths in indigenous and exotic c hic k ens in Vietnam : association of the in tensit y of infection with the Ma jo r Histocompatibility Complex. Par asitolo gy , 134 , 561- 573. [9] Sc hou, T.L.H, Lab ouriau, R., P ermin, A., Christensen, J.P ., Sør ensen, P ., Cu, H.P ., Nguy en, K. N. and Juul-Madsen, H.R. (20 08). MHC hap- lot yp e and susceptibilit y to exp erimen tal infections ( Salmonel la Enter- itidis , Pasteur el la multo cida or A sc aridia ga l li ) in a commercial a nd an indigenous c hic k en breed. P ap er I I I in the PhD thesis “G ene tic dive r- sit y of p oultry in extensiv e pro duction systems a nd its implications for disease resistance” by T o rben Wilde Sc hou, Departmen t of V eterinary P athobiology , F acult y o f L ife Sciences, Univ ersit y of Cop enhagen, Den- mark (submitted).
Original Paper
Loading high-quality paper...
Comments & Academic Discussion
Loading comments...
Leave a Comment