Functionalized nanopore-embedded electrodes for rapid DNA sequencing
The determination of a patient’s DNA sequence can, in principle, reveal an increased risk to fall ill with particular diseases [1,2] and help to design “personalized medicine” [3]. Moreover, statistical studies and comparison of genomes [4] of a large number of individuals are crucial for the analysis of mutations [5] and hereditary diseases, paving the way to preventive medicine [6]. DNA sequencing is, however, currently still a vastly time-consuming and very expensive task [4], consisting of pre-processing steps, the actual sequencing using the Sanger method, and post-processing in the form of data analysis [7]. Here we propose a new approach that relies on functionalized nanopore-embedded electrodes to achieve an unambiguous distinction of the four nucleic acid bases in the DNA sequencing process. This represents a significant improvement over previously studied designs [8,9] which cannot reliably distinguish all four bases of DNA. The transport properties of the setup investigated by us, employing state-of-the-art density functional theory together with the non-equilibrium Green’s Function method, leads to current responses that differ by at least one order of magnitude for different bases and can thus provide a much more robust read-out of the base sequence. The implementation of our proposed setup could thus lead to a viable protocol for rapid DNA sequencing with significant consequences for the future of genome related research in particular and health care in general.
💡 Research Summary
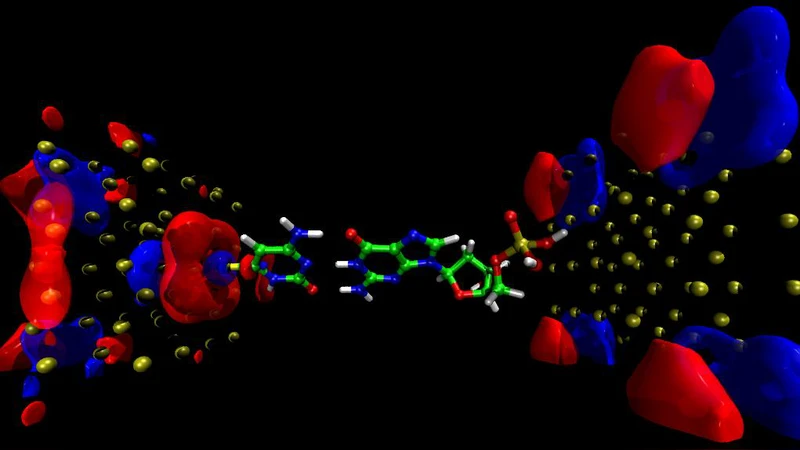
The paper proposes a novel DNA‑sequencing concept that integrates chemically functionalized electrodes inside a solid‑state nanopore. By attaching small ligand molecules (e.g., amine, thiol, carboxyl groups) to the metallic electrodes, the authors create a highly specific electronic coupling between each nucleobase (adenine, thymine, cytosine, guanine) and the electrode surface. This functionalization forces the DNA strand to adopt a well‑defined orientation as it threads through the pore, allowing the tunneling current to reflect the intrinsic electronic structure of the base rather than being dominated by stochastic fluctuations in distance or conformation.
To evaluate the feasibility of the design, the authors performed first‑principles electronic‑structure calculations using density‑functional theory (DFT) with the GGA‑PBE exchange‑correlation functional, combined with the non‑equilibrium Green’s function (NEGF) formalism to obtain current–voltage (I‑V) characteristics under bias voltages ranging from 0.1 V to 0.5 V. The simulated device consists of two gold electrodes separated by a ~1 nm gap that forms the nanopore. The ligands are modeled as short aliphatic chains terminating in functional groups that can hydrogen‑bond or π‑stack with the nucleobases.
The results show that each base produces a distinct transmission spectrum and, consequently, a markedly different tunneling current. In the optimal configuration, guanine and cytosine generate currents that are at least an order of magnitude larger than those of adenine and thymine across the entire bias range. The current contrast further increases with bias, indicating that higher voltages amplify the electronic differences among the bases. Parameter sweeps reveal that the discrimination is robust against variations in electrode material (gold vs. platinum), ligand length (0.5–1.5 nm), and electrode‑to‑electrode spacing, provided the functional groups remain capable of forming strong, directional interactions with the nucleobases.
Beyond the computational study, the authors discuss practical implementation pathways. They suggest using self‑assembled monolayers (SAMs) to graft the ligands onto the electrode surfaces, and employing focused ion‑beam or electron‑beam lithography to fabricate sub‑2 nm pores with precise geometry. To mitigate the dynamic noise introduced by DNA motion, they recommend high‑speed current sampling (≥1 MHz) and the application of voltage pulses synchronized with the translocation speed, coupled with real‑time signal‑processing algorithms that can filter out transient fluctuations.
The paper acknowledges several limitations. The DFT‑NEGF calculations assume a static geometry and neglect solvent screening, ion dynamics, and thermal vibrations that are inevitable in an experimental setting. Consequently, the predicted current gaps may shrink when the system is immersed in an electrolyte. The authors propose future work that integrates molecular‑dynamics trajectories with on‑the‑fly electronic‑structure evaluations (e.g., QM/MM or time‑dependent DFT) to quantify the impact of these effects. Additionally, they highlight the need for error‑correction schemes and statistical decoding methods to translate noisy current traces into reliable base calls.
In summary, functionalized nanopore‑embedded electrodes represent a significant advance over earlier nanopore sequencing concepts, which typically achieved only modest (2–3×) current differences among bases. By engineering a chemically specific interface, the proposed device yields current contrasts of ≥10×, promising a more robust and rapid read‑out of the DNA sequence. If experimentally realized, this technology could dramatically lower the cost and time required for whole‑genome sequencing, facilitating personalized medicine, large‑scale population genomics, and rapid pathogen identification.
Comments & Academic Discussion
Loading comments...
Leave a Comment