Evolution of complex modular biological networks
Biological networks have evolved to be highly functional within uncertain environments while remaining extremely adaptable. One of the main contributors to the robustness and evolvability of biological networks is believed to be their modularity of function, with modules defined as sets of genes that are strongly interconnected but whose function is separable from those of other modules. Here, we investigate the in silico evolution of modularity and robustness in complex artificial metabolic networks that encode an increasing amount of information about their environment while acquiring ubiquitous features of biological, social, and engineering networks, such as scale-free edge distribution, small-world property, and fault-tolerance. These networks evolve in environments that differ in their predictability, and allow us to study modularity from topological, information-theoretic, and gene-epistatic points of view using new tools that do not depend on any preconceived notion of modularity. We find that for our evolved complex networks as well as for the yeast protein-protein interaction network, synthetic lethal pairs consist mostly of redundant genes that lie close to each other and therefore within modules, while knockdown suppressor pairs are farther apart and often straddle modules, suggesting that knockdown rescue is mediated by alternative pathways or modules. The combination of network modularity tools together with genetic interaction data constitutes a powerful approach to study and dissect the role of modularity in the evolution and function of biological networks.
💡 Research Summary
The paper investigates how modularity and robustness emerge in complex biological networks by evolving artificial metabolic networks in silico. The authors construct a population of networks composed of enzymes (nodes) and metabolic reactions (edges), each enzyme being encoded by a gene whose expression can be regulated by environmental cues. Three classes of environments are defined based on predictability: high, intermediate, and low. Networks evolve over many generations under a fitness function that combines product yield, energetic efficiency, and responsiveness to environmental change.
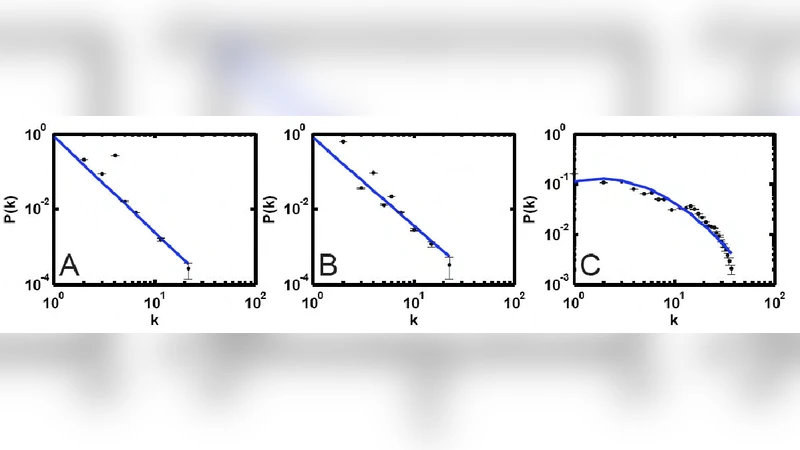
During evolution, the networks spontaneously acquire hallmark features of many real‑world systems: a scale‑free degree distribution, a small‑world topology with high clustering and short average path length, and fault‑tolerance that is especially pronounced when hub nodes are preserved. To quantify modularity without imposing a preconceived community definition, the authors introduce two complementary metrics. The first, an Information‑Flow Module (IFM) measure, evaluates the probability that a random walk starting at one node reaches another, thereby identifying subgraphs that efficiently transmit information. The second, an epistatic interaction matrix, captures genetic interaction strengths; high positive epistasis within a subgraph signals functional redundancy, whereas low or negative epistasis across subgraphs indicates separable functions. By integrating IFM and epistasis, the authors obtain a nuanced picture of both intra‑module cohesion and inter‑module independence.
The results reveal a clear dependence of network architecture on environmental predictability. In highly predictable settings, modules become sharply delineated, each specializing in processing a particular environmental signal or metabolite. Conversely, under low predictability, inter‑module connections proliferate, creating multiple alternative pathways that buffer the system against sudden changes. This shift from tightly compartmentalized to more interwoven structures explains how the same network can be both robust (resistant to random failures) and evolvable (capable of generating novel phenotypes).
To validate the computational findings, the authors map their analyses onto the yeast (Saccharomyces cerevisiae) protein‑protein interaction (PPI) network, using published synthetic lethal (SL) and knock‑down suppressor (KDS) interaction datasets. They find that most SL pairs are located within the same IFM and exhibit strong positive epistasis, reflecting functional redundancy that makes simultaneous loss lethal. In contrast, KDS pairs tend to span different modules or sit at module boundaries, and they display weaker epistatic signals, suggesting that rescue occurs through activation of alternative pathways residing in separate modules. This correspondence demonstrates that the artificial evolution framework captures biologically relevant modular organization and genetic interaction patterns.
Overall, the study provides three major insights: (1) environmental predictability shapes the emergence of modular versus inter‑connected network topologies; (2) information‑flow‑based modularity metrics combined with epistatic data can objectively identify functional modules without prior assumptions; (3) the structural placement of gene pairs predicts the nature of their genetic interactions—redundant, intra‑module pairs give rise to synthetic lethality, while inter‑module pairs underlie knock‑down suppression. By showing that artificially evolved networks recapitulate key properties of real biological systems, the work offers a powerful platform for probing the principles that allow complex networks to be simultaneously robust and adaptable, with implications for synthetic biology, evolutionary theory, and the design of resilient engineered systems.
Comments & Academic Discussion
Loading comments...
Leave a Comment