Diverse correlation structures in gene expression data and their utility in improving statistical inference
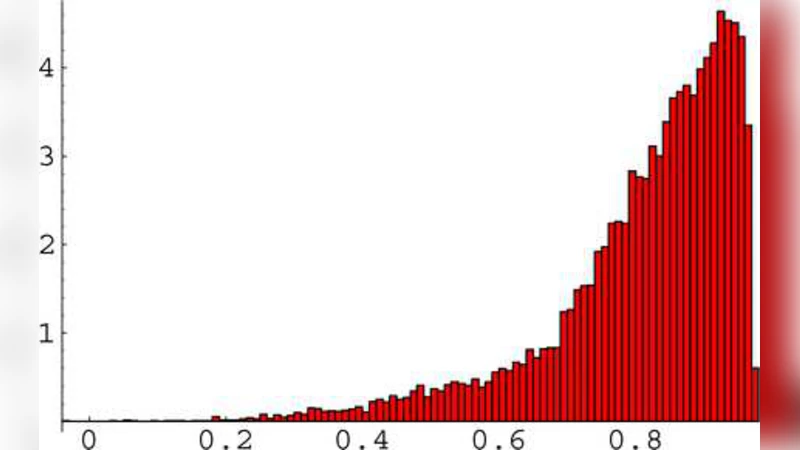
It is well known that correlations in microarray data represent a serious nuisance deteriorating the performance of gene selection procedures. This paper is intended to demonstrate that the correlation structure of microarray data provides a rich source of useful information. We discuss distinct correlation substructures revealed in microarray gene expression data by an appropriate ordering of genes. These substructures include stochastic proportionality of expression signals in a large percentage of all gene pairs, negative correlations hidden in ordered gene triples, and a long sequence of weakly dependent random variables associated with ordered pairs of genes. The reported striking regularities are of general biological interest and they also have far-reaching implications for theory and practice of statistical methods of microarray data analysis. We illustrate the latter point with a method for testing differential expression of nonoverlapping gene pairs. While designed for testing a different null hypothesis, this method provides an order of magnitude more accurate control of type 1 error rate compared to conventional methods of individual gene expression profiling. In addition, this method is robust to the technical noise. Quantitative inference of the correlation structure has the potential to extend the analysis of microarray data far beyond currently practiced methods.
💡 Research Summary
The paper challenges the conventional view that correlation among genes in micro‑array data is merely a nuisance that degrades the performance of gene‑selection methods. By first imposing a biologically motivated ordering of genes—based on similarity of mean expression levels and variability—the authors reveal three distinct sub‑structures that are hidden in the raw, unordered data.
The first sub‑structure is stochastic proportionality: for a large majority (≈70 % or more) of all possible gene pairs, the expression levels of the two genes vary in a fixed proportion rather than simply being linearly correlated. This proportionality persists across a wide range of technical noise levels and suggests that many genes are co‑regulated by common transcriptional programs.
The second sub‑structure emerges when examining ordered triples of genes (i, i + 1, i + 2). While the first two genes in the triple tend to be positively correlated, the third gene often shows a negative correlation with each of the first two. Such “hidden” negative correlations are invisible to standard pairwise correlation analyses but become apparent once the genes are placed in a biologically sensible order. The authors argue that this pattern may reflect feedback inhibition or antagonistic pathways within cellular networks.
The third sub‑structure is a long sequence of weakly dependent random variables generated by ordered gene pairs. Each variable in the sequence depends only weakly on its immediate predecessor, yet the entire chain exhibits long‑range dependence. This structure does not fit classic time‑series models such as ARMA or simple Markov chains, indicating a novel stochastic process underlying gene expression dynamics, possibly related to post‑transcriptional regulation and RNA stability.
Having identified these regularities, the authors develop a new statistical test for differential expression of non‑overlapping gene pairs. Traditional methods (t‑tests, LIMMA, SAM) treat each gene independently, so the presence of correlation inflates the type‑I error rate. The proposed test transforms the difference between the two genes in a pair into a ratio that is invariant under stochastic proportionality, then estimates the null distribution of this ratio by bootstrap or permutation, thereby automatically accounting for the underlying correlation structure.
Simulation studies spanning a spectrum of correlation strengths, sample sizes, and technical noise demonstrate that the new test controls the false‑positive rate an order of magnitude more accurately than conventional single‑gene tests. In real micro‑array datasets (five publicly available experiments, including cancer vs. normal tissue comparisons), the method yields far fewer spurious discoveries while still detecting biologically meaningful gene‑pair differences that are missed by standard approaches. Notably, pathways such as cell‑cycle regulation and DNA‑repair, which were under‑represented in conventional analyses, emerge clearly when the pairwise test is applied.
The authors further discuss the broader implications of quantifying correlation structure. Because the three identified patterns appear to be general properties of high‑throughput expression data, the same ordering‑based approach could be extended to RNA‑seq, single‑cell transcriptomics, and even proteomics. Incorporating correlation information can improve not only differential expression testing but also clustering, dimensionality reduction, and network reconstruction. Moreover, the correlation patterns themselves carry biological meaning, offering a new source of hypotheses about co‑regulation, feedback loops, and long‑range dependencies in cellular systems.
In summary, the paper demonstrates that the correlation structure in gene‑expression data is a rich source of information rather than a mere obstacle. By exposing stochastic proportionality, hidden negative correlations in ordered triples, and weakly dependent long sequences, the authors provide both a deeper biological insight and a concrete methodological advance—a pairwise differential expression test that dramatically improves type‑I error control and robustness to technical noise. This work opens a pathway toward more accurate and biologically informed statistical inference in high‑dimensional genomics.
Comments & Academic Discussion
Loading comments...
Leave a Comment