Computational analysis of folding and mutation properties of C5 domain from Myosin binding protein C
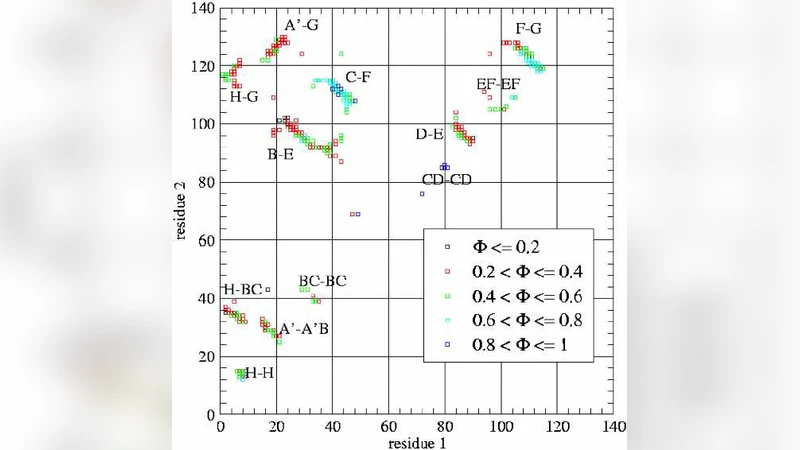
Thermal folding Molecular Dynamics simulations of the domain C5 from Myosin Binding Protein C were performed using a native-centric model to study the role of three mutations related to Familial Hypertrophic Cardiomyopathy. Mutation of Asn755 causes the largest shift of the folding temperature, and the residue is located in the CFGA’ beta-sheet featuring the highest Phi-values. The mutation thus appears to reduce the thermodynamic stability in agreement with experimental data. The mutations on Arg654 and Arg668, conversely, cause a little change in the folding temperature and they reside in the low Phi-value BDE beta-sheet, so that their pathologic role cannot be related to impairment of the folding process but possibly to the binding with target molecules. As the typical signature of Domain C5 is the presence of a longer and destabilizing CD-loop with respect to the other Ig-like domains we completed the work with a bioinformatic analysis of this loop showing a high density of negative charge and low hydrophobicity. This indicates the CD-loop as a natively unfolded sequence with a likely coupling between folding and ligand binding.
💡 Research Summary
The paper presents a combined computational and bioinformatic investigation of the C5 Ig‑like domain of Myosin Binding Protein C (MyBP‑C), focusing on its thermal folding behavior and the impact of three Familial Hypertrophic Cardiomyopathy (FHC)–associated point mutations. Using a native‑centric coarse‑grained potential, the authors performed temperature‑gradient molecular dynamics simulations for the wild‑type domain and for three mutant variants: Asn755, Arg654, and Arg668. For each trajectory they extracted the folding transition temperature (Tm) and calculated Φ‑values, which quantify the degree to which individual residues are structured in the transition state ensemble.
The results reveal a clear dichotomy among the mutations. Asn755 resides in the CFGA′ β‑sheet, a region that exhibits the highest Φ‑values (≈0.8–0.9), indicating that it is a critical nucleus for domain folding. The Asn→X substitution lowers the folding temperature by roughly 5 °C, a substantial destabilization that aligns with experimental observations of reduced thermodynamic stability in the corresponding patient‑derived protein. By contrast, Arg654 and Arg668 are located in the BDE β‑sheet, a segment characterized by low Φ‑values (≈0.2–0.3). Their mutations produce only marginal shifts in Tm (≤0.5 °C), suggesting that they do not appreciably perturb the folding pathway. The authors therefore propose that the pathogenic effect of these arginine residues is more likely related to altered binding interactions with target molecules, such as actin‑myosin complexes, rather than to a folding defect.
In addition to the folding analysis, the study includes a detailed sequence‑based characterization of the C5 domain’s CD‑loop, which is unusually long compared with other Ig‑like domains in MyBP‑C. Using charge distribution calculations and Kyte‑Doolittle hydropathy profiling, the authors demonstrate that the loop is enriched in negatively charged residues (Asp/Glu >45 % of the loop) and has a low average hydrophobicity (mean hydropathy ≈ –1.2). These features are hallmarks of intrinsically disordered regions (IDRs). The authors argue that the CD‑loop therefore exists in a natively unfolded state under physiological conditions and may undergo disorder‑to‑order transitions upon binding to specific ligands, coupling folding to functional interaction.
Overall, the work showcases the utility of native‑centric molecular dynamics for dissecting the thermodynamic consequences of disease‑related mutations at a residue‑specific level. It also highlights how sequence‑derived biophysical parameters can identify functional disorder within a protein domain. By integrating these approaches, the authors provide a mechanistic framework that distinguishes mutations that destabilize the C5 domain’s native fold (e.g., Asn755) from those that likely impair protein‑protein interactions without affecting global stability (e.g., Arg654, Arg668). This distinction has implications for therapeutic strategies: stabilizing the folded state may be beneficial for mutations like Asn755, whereas targeting the interaction interface could be more appropriate for the arginine variants. The identification of the CD‑loop as a potential disorder‑mediated binding platform further expands the functional repertoire of MyBP‑C and suggests new avenues for experimental validation and drug design.
Comments & Academic Discussion
Loading comments...
Leave a Comment