Emergence of clonal selection and affinity maturation in an ab initio microscopic model of immunity
Mechanisms of immunity, and of the host-pathogen interactions in general are among the most fundamental problems of medicine, ecology, and evolution studies. Here, we present a microscopic, protein-level, sequence-based model of immune system, with explicitly defined interactions between host and pathogen proteins.. Simulations of this model show that possible outcomes of the infection (extinction of cells, survival with complete elimination of viruses, or chronic infection with continuous coexistence of cells and viruses) crucially depend on mutation rates of the viral and immunoglobulin proteins. Infection is always lethal if the virus mutation rate exceeds a certain threshold. Potent immunoglobulins are discovered in this model via clonal selection and affinity maturation. Surviving cells acquire lasting immunity against subsequent infection by the same virus strain. As a second line of defense cells develop apoptosis-like behavior by reducing their lifetimes to eliminate viruses. These results demonstrate the feasibility of microscopic sequence-based models of immune system, where population dynamics of the evolving B-cells is explicitly tied to the molecular properties of their proteins.
💡 Research Summary
The paper introduces a bottom‑up, protein‑level, sequence‑based computational model of the adaptive immune response, focusing on the interaction between host B‑cell immunoglobulins (Ig) and viral surface proteins. Each B‑cell is represented by a single Ig gene, and each virus by a surface protein gene; both evolve through point mutations. The binding affinity between an Ig and a viral protein is calculated from a physics‑inspired energy function that accounts for hydrophobic contacts, hydrogen bonds, and electrostatic interactions. This affinity directly modulates the cell’s replication probability and death rate, thereby linking molecular properties to population dynamics.
The simulation proceeds in discrete time steps. At each step the virus replicates, mutates, and the current pool of Ig‑producing B‑cells also mutates. For every possible Ig–virus pair the model computes a binding energy; cells whose Ig exhibits higher affinity enjoy increased proliferation, while low‑affinity cells are more likely to die. Selected high‑affinity clones expand (clonal selection) and subsequently undergo further rounds of mutation and selection, mimicking affinity maturation observed in germinal centers. The authors systematically vary two key parameters: the viral mutation rate (µ_v) and the Ig mutation rate (µ_Ig), while keeping other factors (viral replication rate, baseline cell death, etc.) constant.
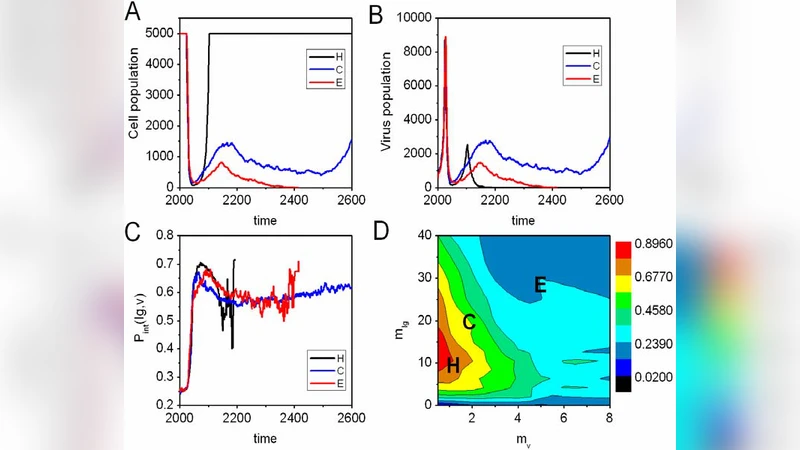
Three qualitatively distinct infection outcomes emerge:
-
Lethal infection – When µ_v exceeds a critical threshold (≈10⁻³ per residue per replication in the authors’ parameterization), the virus explores sequence space faster than the Ig repertoire can adapt. High‑affinity Ig clones never arise before the viral load overwhelms the host, leading to complete cell extinction. This regime mirrors high‑mutation RNA viruses such as influenza or HIV, where immune escape can be fatal.
-
Successful clearance via clonal selection and affinity maturation – For intermediate µ_v values, a subset of randomly generated Ig clones initially binds the virus weakly. Those clones are preferentially amplified; subsequent rounds of mutation (somatic hypermutation) and selection increase their binding energy logarithmically over time. The model reproduces the classic “affinity maturation curve,” showing a steady rise in average Ig affinity and a concomitant drop in viral load until the virus is eradicated. Surviving cells retain the high‑affinity Ig genes, providing durable immunity against re‑infection with the same viral strain.
-
Chronic coexistence – At low µ_v, the virus mutates slowly, allowing a quasi‑steady state where both virus and host cells persist. In this regime the model introduces a secondary defense: infected cells shorten their intrinsic lifespan (programmed cell death), effectively limiting the time window for viral replication within each cell. This apoptosis‑like response reduces the overall viral burden without eliminating the pathogen, reminiscent of chronic infections where host tolerance mechanisms dominate.
The authors also demonstrate immune memory: after clearance, the high‑affinity Ig clones remain in the population, and a subsequent exposure to the identical virus triggers an immediate, robust response, preventing re‑establishment of infection.
Methodologically, the study employs a Monte‑Carlo evolutionary algorithm. Each iteration samples mutations according to the specified rates, evaluates binding energies using a coarse‑grained potential, and applies selection based on fitness functions derived from those energies. Parameter sweeps identify the critical viral mutation rate that separates lethal from survivable infections. The model’s simplicity allows exhaustive exploration of the genotype‑phenotype‑population map, something infeasible in full‑scale agent‑based or differential‑equation models.
Limitations are acknowledged. The model omits T‑cell help, cytokine signaling, antigen presentation, and the spatial organization of germinal centers, all of which are known to shape real‑world affinity maturation. Viral replication dynamics are reduced to a single fitness term, ignoring mechanisms such as latency, recombination, or immune evasion strategies beyond point mutation. Moreover, the binding energy function relies on approximate structural predictions; inaccuracies could affect the quantitative thresholds reported.
Despite these simplifications, the work convincingly shows that a sequence‑level, physics‑based description can reproduce hallmark features of adaptive immunity: clonal selection, affinity maturation, immune memory, and a mutation‑rate‑dependent phase diagram of infection outcomes. It opens a pathway toward multi‑scale immunological modeling where molecular evolution feeds directly into epidemiological predictions, and suggests future extensions incorporating T‑cell dynamics, more realistic viral life cycles, and experimentally calibrated interaction potentials.
Comments & Academic Discussion
Loading comments...
Leave a Comment