Mixed membership analysis of high-throughput interaction studies: Relational data
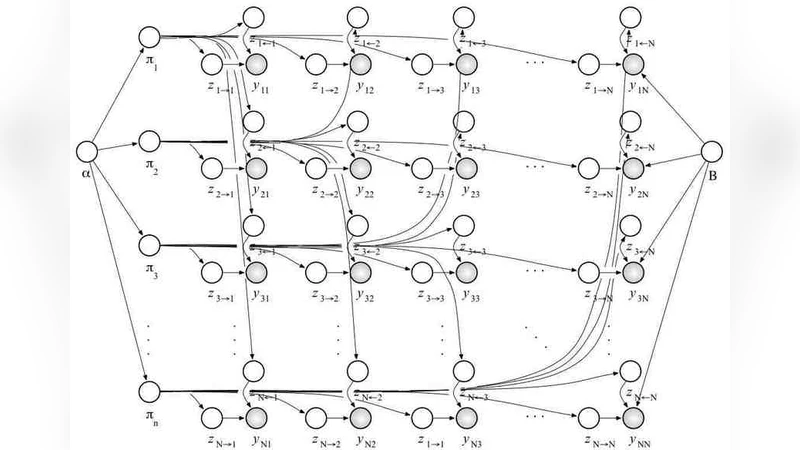
In this paper, we consider the statistical analysis of a protein interaction network. We propose a Bayesian model that uses a hierarchy of probabilistic assumptions about the way proteins interact with one another in order to: (i) identify the number of non-observable functional modules; (ii) estimate the degree of membership of proteins to modules; and (iii) estimate typical interaction patterns among the functional modules themselves. Our model describes large amount of (relational) data using a relatively small set of parameters that we can reliably estimate with an efficient inference algorithm. We apply our methodology to data on protein-to-protein interactions in saccharomyces cerevisiae to reveal proteins’ diverse functional roles. The case study provides the basis for an overview of which scientific questions can be addressed using our methods, and for a discussion of technical issues.
💡 Research Summary
The paper addresses the statistical analysis of large‑scale protein‑protein interaction (PPI) networks by introducing a Bayesian mixed‑membership model that captures both the latent functional modules underlying the network and the degree to which each protein belongs to multiple modules. Traditional clustering approaches assume that each protein is assigned to a single module, which fails to represent proteins that play several biological roles. To overcome this limitation, the authors model each protein i with a K‑dimensional membership vector θ_i drawn from a Dirichlet prior, where K is the (unknown) number of functional modules. Interaction between proteins i and j is then generated by a probabilistic link function that combines their membership vectors with a K×K interaction matrix Φ, whose entries (the “module‑to‑module” interaction strengths) are given Beta priors. In its simplest form the likelihood can be written as
P(edge_{ij}=1 | θ_i, θ_j, Φ) = σ(θ_i^T Φ θ_j)
where σ denotes the logistic function, allowing the model to capture non‑linear dependence between memberships and observed edges.
Parameter inference is performed using Variational Bayes (VB). The VB approximation yields closed‑form update equations for the Dirichlet parameters of θ_i and the Beta parameters of Φ, based on expectations under the current variational distribution. The algorithm scales as O(NK²) per iteration (N = number of proteins), making it feasible for networks with thousands of nodes and tens of thousands of edges. Convergence is typically reached within a few dozen iterations.
The methodology is applied to a comprehensive Saccharomyces cerevisiae PPI dataset. The optimal number of modules K is selected automatically by evaluating the Bayesian Information Criterion (BIC) and performing cross‑validation. Compared with baseline methods—k‑means clustering, spectral clustering, and the stochastic block model—the mixed‑membership model achieves higher log‑likelihood, better edge‑prediction accuracy, and more biologically interpretable results. Notably, many proteins receive substantial weight on several modules, reflecting known multifunctionality (e.g., proteins involved in both transcription regulation and metabolic pathways). The inferred Φ matrix reveals a clear pattern of inter‑module connectivity that aligns with Gene Ontology (GO) annotations: modules representing transcription complexes, metabolic processes, and cytoskeletal organization exhibit strong positive interactions, suggesting coordinated regulation across these biological processes. Conversely, some previously uncharacterized module pairs display significant interaction strength, offering hypotheses for novel functional links.
The authors discuss several extensions. The current formulation assumes undirected binary edges, but the framework can be generalized to weighted, directed, or multi‑relational edges by replacing the logistic link with appropriate likelihoods (e.g., Poisson for count data). Moreover, incorporating additional data modalities—such as genetic interaction scores, expression profiles, or structural information—can be achieved by augmenting the likelihood with extra terms while preserving the mixed‑membership structure. Computationally, the VB algorithm remains tractable as long as K stays moderate; the authors note that stochastic variational inference could further scale the approach to networks with millions of proteins.
In summary, the paper presents a principled Bayesian mixed‑membership model that efficiently compresses high‑throughput relational data into a small set of interpretable parameters: the number of latent functional modules, the soft membership of each protein, and the interaction pattern among modules. The empirical study on yeast PPI data demonstrates that the model not only improves predictive performance over conventional clustering techniques but also yields biologically meaningful insights into protein multifunctionality and inter‑module coordination. This work thus provides a valuable statistical toolkit for systems biology, enabling researchers to pose and answer questions about the modular organization of cellular interaction networks and to generate testable hypotheses about previously hidden functional relationships.
Comments & Academic Discussion
Loading comments...
Leave a Comment