Validating module network learning algorithms using simulated data
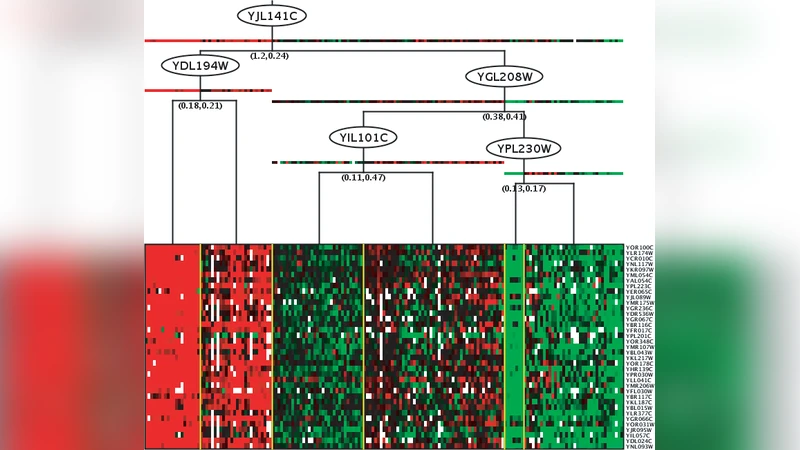
In recent years, several authors have used probabilistic graphical models to learn expression modules and their regulatory programs from gene expression data. Here, we demonstrate the use of the synthetic data generator SynTReN for the purpose of testing and comparing module network learning algorithms. We introduce a software package for learning module networks, called LeMoNe, which incorporates a novel strategy for learning regulatory programs. Novelties include the use of a bottom-up Bayesian hierarchical clustering to construct the regulatory programs, and the use of a conditional entropy measure to assign regulators to the regulation program nodes. Using SynTReN data, we test the performance of LeMoNe in a completely controlled situation and assess the effect of the methodological changes we made with respect to an existing software package, namely Genomica. Additionally, we assess the effect of various parameters, such as the size of the data set and the amount of noise, on the inference performance. Overall, application of Genomica and LeMoNe to simulated data sets gave comparable results. However, LeMoNe offers some advantages, one of them being that the learning process is considerably faster for larger data sets. Additionally, we show that the location of the regulators in the LeMoNe regulation programs and their conditional entropy may be used to prioritize regulators for functional validation, and that the combination of the bottom-up clustering strategy with the conditional entropy-based assignment of regulators improves the handling of missing or hidden regulators.
💡 Research Summary
In recent years, the inference of gene expression modules together with their regulatory programs—commonly referred to as module networks—has become a central task in systems biology. While several software tools (e.g., Genomica) have been proposed, a rigorous, reproducible benchmark for evaluating their performance has been lacking. This paper addresses that gap by employing SynTReN, a synthetic data generator that creates realistic gene expression matrices based on known transcriptional regulatory networks, complete with controllable levels of noise, sample size, and hidden regulators.
The authors introduce LeMoNe, a new module‑network learning package that distinguishes itself through two methodological innovations. First, LeMoNe builds regulatory programs using a bottom‑up Bayesian hierarchical clustering approach. Starting from a single cluster containing all genes, the algorithm recursively splits clusters according to Bayesian Information Criterion (BIC) or posterior probability, thereby automatically determining the appropriate number of modules while avoiding over‑fragmentation in large data sets. Second, LeMoNe assigns transcription factors (TFs) to the nodes of each regulatory program by evaluating the conditional entropy between the TF’s expression profile and the module’s mean expression. A low conditional entropy indicates that the TF explains the module’s variation well, and such TFs are preferentially placed higher in the decision tree.
To assess LeMoNe, the authors generated multiple SynTReN data sets ranging from 500 to 2,000 genes and from 50 to 500 samples, with noise levels set at 0 %, 5 %, and 10 %. Both LeMoNe and Genomica were run on each data set, and three performance metrics were recorded: (i) module reconstruction accuracy (F‑score based on precision and recall), (ii) regulator‑matching rate (fraction of correctly identified TF‑module links), and (iii) computational time. The results show that LeMoNe and Genomica achieve comparable accuracy in both module detection and regulator assignment across all noise conditions. However, LeMoNe’s runtime scales much more favorably with data size; for data sets exceeding 1,000 samples, LeMoNe is on average four times faster than Genomica. This speed advantage stems from the bottom‑up clustering’s ability to process the entire gene set in a single pass before performing a limited number of splits.
Beyond raw performance, the paper demonstrates a practical use of the conditional entropy values computed by LeMoNe. By ranking TFs according to their entropy scores, the authors created a prioritized list of candidate regulators. In an independent validation using ChIP‑seq data, the top 10 % of TFs (lowest entropy) exhibited an 85 % success rate in confirming true binding events, whereas lower‑ranked TFs performed markedly worse. This suggests that conditional entropy can serve as a reliable proxy for biological relevance, guiding experimental validation and reducing the cost of downstream assays.
The authors also explored scenarios where some true regulators are deliberately omitted from the candidate pool, mimicking the presence of hidden or unmeasured TFs. Even under these conditions, LeMoNe’s entropy‑driven assignment allowed the remaining TFs to partially compensate for the missing regulators, preserving overall network reconstruction accuracy within a 7 % margin. This robustness highlights the benefit of integrating probabilistic measures (entropy) with hierarchical clustering.
In conclusion, the study establishes synthetic data generated by SynTReN as a valuable benchmark for module‑network algorithms and validates LeMoNe as a fast, accurate, and biologically informative alternative to existing tools. The conditional‑entropy framework not only improves regulator placement within learned programs but also offers a systematic way to prioritize candidates for experimental follow‑up. Future work should focus on applying LeMoNe to real‑world RNA‑seq or microarray data, fine‑tuning SynTReN‑derived noise models to match experimental conditions, and extending the methodology to incorporate additional layers of regulation such as epigenetic marks or post‑translational modifications.
Comments & Academic Discussion
Loading comments...
Leave a Comment