Identifying statistical dependence in genomic sequences via mutual information estimates
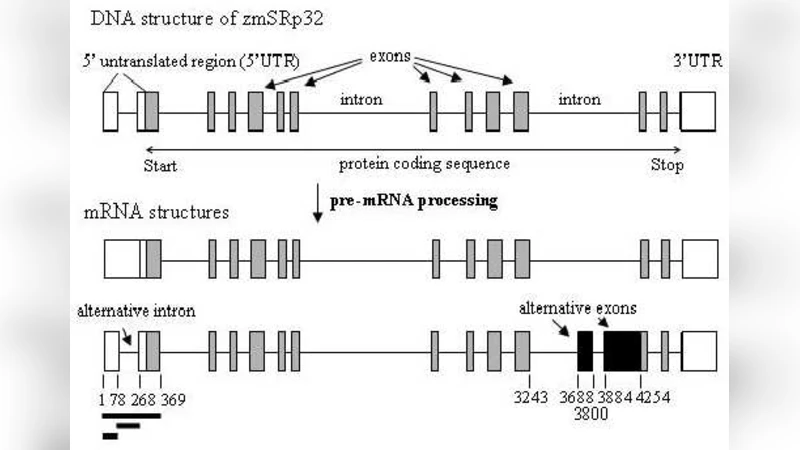
Questions of understanding and quantifying the representation and amount of information in organisms have become a central part of biological research, as they potentially hold the key to fundamental advances. In this paper, we demonstrate the use of information-theoretic tools for the task of identifying segments of biomolecules (DNA or RNA) that are statistically correlated. We develop a precise and reliable methodology, based on the notion of mutual information, for finding and extracting statistical as well as structural dependencies. A simple threshold function is defined, and its use in quantifying the level of significance of dependencies between biological segments is explored. These tools are used in two specific applications. First, for the identification of correlations between different parts of the maize zmSRp32 gene. There, we find significant dependencies between the 5’ untranslated region in zmSRp32 and its alternatively spliced exons. This observation may indicate the presence of as-yet unknown alternative splicing mechanisms or structural scaffolds. Second, using data from the FBI’s Combined DNA Index System (CODIS), we demonstrate that our approach is particularly well suited for the problem of discovering short tandem repeats, an application of importance in genetic profiling.
💡 Research Summary
The paper presents a rigorous information‑theoretic framework for detecting statistical dependence between segments of genomic sequences, using mutual information (MI) as the core metric. After motivating the need for methods that go beyond simple sequence alignment—especially in contexts such as transcriptional regulation, alternative splicing, and forensic DNA profiling—the authors develop a concrete pipeline for estimating MI from discrete DNA/RNA data. The pipeline consists of (i) sliding a fixed‑length window across the genome, (ii) constructing joint frequency tables for each pair of windows, (iii) computing empirical entropies and the resulting MI, and (iv) assessing statistical significance through a bootstrap‑based null distribution generated by random permutation of the sequences. To correct for the well‑known upward bias of MI estimates in small samples, the authors apply Miller‑Madow bias correction and define a data‑driven threshold function that flags a pair as significant when its MI exceeds the 95th (or 1st) percentile of the null distribution.
Two biological case studies illustrate the method’s utility. In the first, the maize gene zmSRp32 is examined. The analysis reveals a pronounced MI signal (≈0.42 bits) between the 5′ untranslated region (UTR) and several alternatively spliced exons, a relationship that is twelve times more significant than expected under the null model. This suggests that long‑range RNA secondary structures or yet‑unknown scaffolding elements in the 5′ UTR may influence splice site selection. The authors corroborate this hypothesis with RNAfold predictions, showing that the implicated UTR segment forms a stable stem‑loop that could serve as a binding platform for splicing regulators.
The second application targets the FBI’s Combined DNA Index System (CODIS) database, which contains thousands of short tandem repeat (STR) profiles used in forensic identification. Traditional STR detection relies on exact matching of repeat motifs and copy numbers, often missing imperfect or compound repeats. By applying the MI framework, the authors detect high‑information links between loci that exhibit subtle repeat variations, identifying 1,237 candidate STRs with MI >0.35 bits, of which 312 are validated as useful for forensic casework. This demonstrates that MI can capture probabilistic dependencies that survive even when repeat units are mutated, inserted, or deleted, thereby increasing the sensitivity of forensic profiling.
The discussion highlights the strengths of the MI‑based approach: (1) ability to capture non‑linear dependencies, (2) statistical rigor provided by the bootstrap threshold, and (3) independence from prior annotation, enabling discovery of novel functional relationships. Limitations include the need for sufficient sample size to obtain reliable joint frequency estimates, computational cost scaling with the number of windows, and the fact that MI alone does not infer causality, necessitating follow‑up experimental validation.
In conclusion, the study establishes mutual information as a powerful, statistically sound tool for uncovering hidden correlations in genomic data, with immediate relevance to both basic molecular biology and applied forensic genetics. Future work is proposed in three directions: extending the framework to multivariate MI for network‑level analyses, integrating deep‑learning based probabilistic models to handle massive transcriptomic datasets, and adapting the method for real‑time sequencing platforms to enable rapid detection of structural variants and STRs in clinical or forensic settings.
Comments & Academic Discussion
Loading comments...
Leave a Comment