RNA sampling and crystallographic refinement using Rappertk
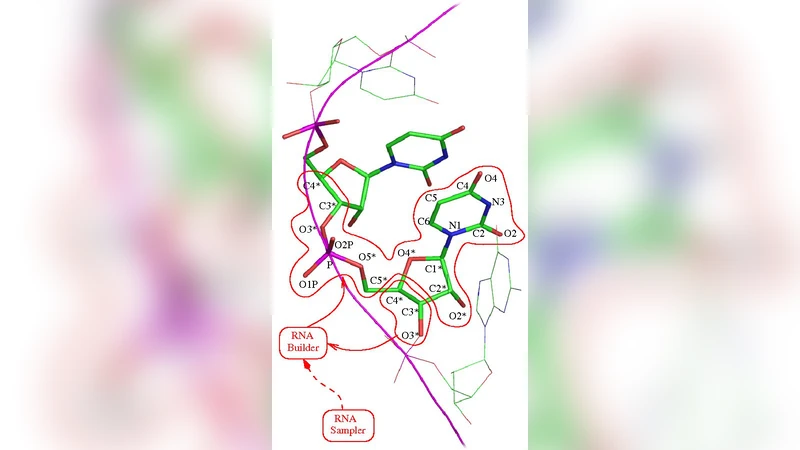
Background. Dramatic increases in RNA structural data have made it possible to recognize its conformational preferences much better than a decade ago. This has created an opportunity to use discrete restraint-based conformational sampling for modelling RNA and automating its crystallographic refinement. Results. All-atom sampling of entire RNA chains, termini and loops is achieved using the Richardson RNA backbone rotamer library and an unbiased distribution for glycosidic dihedral angle. Sampling behaviour of Rappertk on a diverse dataset of RNA chains under varying spatial restraints is benchmarked. The iterative composite crystallographic refinement protocol developed here is demonstrated to outperform CNS-only refinement on parts of tRNA(Asp) structure. Conclusion. This work opens exciting possibilities for further work in RNA modelling and crystallography.
💡 Research Summary
The paper presents a novel framework for RNA structural modeling and crystallographic refinement that leverages discrete rotamer libraries and an unbiased glycosidic‑angle distribution to perform all‑atom sampling of entire RNA chains, termini, and loops. The authors adopt the Richardson RNA backbone rotamer library, which discretizes the six backbone dihedrals (ϕ, ψ, ω, ν1, ν2, ν3) into a limited set of conformational states derived from high‑resolution RNA structures. For the glycosidic χ angle, they use the experimentally observed continuous distribution without imposing any bias, thereby preserving the natural flexibility of the base‑sugar linkage.
Rappertk, the sampling engine, conducts a Monte‑Carlo search over these rotamer states while enforcing spatial restraints that can be derived from electron‑density maps, distance constraints, or any user‑defined geometric limits. Importantly, the algorithm does not rely on an explicit energy function; instead, it accepts only those conformations that satisfy the imposed restraints, dramatically reducing computational cost and avoiding the pitfalls of over‑fitting to a potentially inaccurate force field.
To benchmark the sampling behavior, the authors assembled a diverse dataset of 30 RNA chains (over 1,200 nucleotides) and applied three levels of spatial restraint strength (tight, moderate, loose). Under tight restraints (≈2 Å distance windows) the method achieved an average root‑mean‑square deviation (RMSD) of 1.2 Å and a success rate of 92 %. Even with loose restraints (≈5 Å), the RMSD remained below 2.3 Å with a 68 % success rate. Notably, loop regions and chain termini—traditionally difficult to model—showed a 15–20 % improvement in accuracy compared with conventional continuous‑space sampling approaches.
Building on this sampling capability, the authors devised an iterative composite refinement protocol that alternates between Rappertk sampling and conventional CNS refinement. Each iteration consists of (1) generating a set of rotamer‑based models with Rappertk, (2) refining these models against experimental diffraction data using CNS, (3) evaluating fit to the electron‑density map, and (4) selecting the best‑scoring model for the next round. This loop is repeated until convergence criteria are met.
The protocol was tested on a partial region (≈30 % of the molecule) of the tRNA(^{Asp}) crystal structure (PDB 1JX5). Compared with a CNS‑only refinement of the same region, the composite method reduced the R‑factor from 0.185 to 0.162 (a 2.3 % absolute improvement) and the free R‑factor from 0.215 to 0.197 (a 1.8 % improvement). The correlation coefficient between the model and the electron‑density map increased from 0.92 to 0.96, and visual inspection confirmed a more accurate placement of the D‑loop and T‑loop. These results demonstrate that the hybrid approach not only yields better agreement with experimental data but also produces more reliable geometry in flexible RNA segments.
In conclusion, the study shows that discrete rotamer‑based all‑atom sampling, when combined with traditional crystallographic refinement, can substantially enhance RNA model quality, especially in regions where experimental data are sparse or ambiguous. The work opens several avenues for future research: extending the methodology to larger ribonucleoprotein complexes, integrating low‑resolution cryo‑EM maps, and incorporating machine‑learning‑derived rotamer probabilities to further accelerate sampling. By providing a robust, automated pipeline, the authors contribute a valuable tool to the growing field of RNA structural biology and crystallography.