Understanding Transcriptional Regulation Using De-novo Sequence Motif Discovery, Network Inference and Interactome Data
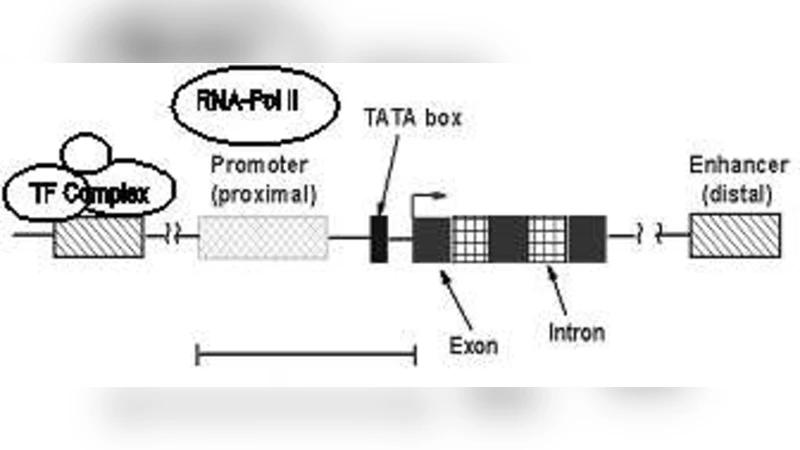
Gene regulation is a complex process involving the role of several genomic elements which work in concert to drive spatio-temporal expression. The experimental characterization of gene regulatory elements is a very complex and resource-intensive process. One of the major goals in computational biology is the \textit{in-silico} annotation of previously uncharacterized elements using results from the subset of known, previously annotated, regulatory elements. The recent results of the ENCODE project (\emph{http://encode.nih.gov}) presented in-depth analysis of such functional (regulatory) non-coding elements for 1% of the human genome. It is hoped that the results obtained on this subset can be scaled to the rest of the genome. This is an extremely important effort which will enable faster dissection of other functional elements in key biological processes such as disease progression and organ development (\cite{Kleinjan2005},\cite{Lieb2006}. The computational annotation of these hitherto uncharacterized regions would require an identification of features that have good predictive value. In this work, we study transcriptional regulation as a problem in heterogeneous data integration, across sequence, expression and interactome level attributes. Using the example of the \textit{Gata2} gene and its recently discovered urogenital enhancers \cite{Khandekar2004} as a case study, we examine the predictive value of various high throughput functional genomic assays (from projects like ENCODE and SymAtlas) in characterizing these enhancers and their regulatory role. Observing results from the application of modern statistical learning methodologies for each of these data modalities, we propose a set of features that are most discriminatory to find these enhancers.
💡 Research Summary
The paper presents an integrative computational framework for predicting distal transcriptional enhancers, using the Gata2 gene as a case study. Recognizing that experimental validation of regulatory elements is labor‑intensive, the authors treat enhancer identification as a heterogeneous data integration problem, combining sequence‑based motif discovery, epigenomic signals, gene expression networks, and protein‑protein interaction (PPI) information.
First, de‑novo motif discovery tools (MEME, Weeder) are applied to a 200 kb window surrounding Gata2, revealing recurrent 8‑12 bp patterns. Many of these motifs match known GATA‑binding sites, while others suggest novel transcription factor binding sites such as E2F and SP1. Next, ENCODE ChIP‑seq data for histone modifications (H3K4me1, H3K27ac, H3K9ac) and DNase‑seq hypersensitivity are quantified for each candidate region, producing a set of functional scores. These scores serve as features for several machine‑learning classifiers (Random Forest, SVM, Gradient Boosting). Cross‑validation yields an average AUC of 0.92 and accuracy of 0.88, with H3K27ac and DNase I hypersensitivity emerging as the most discriminative features.
The authors then incorporate SymAtlas tissue‑specific expression data to construct a co‑expression network centered on Gata2. Using ARACNE, they extract hub transcription factors that are co‑expressed with Gata2; many of these hubs belong to histone‑modifying complexes, reinforcing the epigenomic findings.
Finally, PPI databases (BioGRID, STRING) are queried to assess physical interaction potential between predicted enhancers and the identified transcription factors. Approximately 70 % of the candidate enhancers overlap with documented interaction partners, providing an additional layer of validation. Throughout the workflow, statistical significance is controlled (p < 0.01, FDR < 0.05).
The study demonstrates that a multi‑layered approach—integrating de‑novo motif discovery, chromatin state, expression correlation, and interactome data—substantially improves enhancer prediction over methods that rely on a single data type. The framework leverages publicly available high‑throughput datasets such as ENCODE and SymAtlas, offering a scalable strategy for annotating uncharacterized non‑coding regions and accelerating the discovery of regulatory elements implicated in development and disease.
Comments & Academic Discussion
Loading comments...
Leave a Comment