Probabilistic annotation of protein sequences based on functional classifications
BACKGROUND: One of the most evident achievements of bioinformatics is the development of methods that transfer biological knowledge from characterised proteins to uncharacterised sequences. This mode of protein function assignment is mostly based on the detection of sequence similarity and the premise that functional properties are conserved during evolution. Most automatic approaches developed to date rely on the identification of clusters of homologous proteins and the mapping of new proteins onto these clusters, which are expected to share functional characteristics. RESULTS: Here, we inverse the logic of this process, by considering the mapping of sequences directly to a functional classification instead of mapping functions to a sequence clustering. In this mode, the starting point is a database of labelled proteins according to a functional classification scheme, and the subsequent use of sequence similarity allows defining the membership of new proteins to these functional classes. In this framework, we define the Correspondence Indicators as measures of relationship between sequence and function and further formulate two Bayesian approaches to estimate the probability for a sequence of unknown function to belong to a functional class. This approach allows the parametrisation of different sequence search strategies and provides a direct measure of annotation error rates. We validate this approach with a database of enzymes labelled by their corresponding four-digit EC numbers and analyse specific cases. CONCLUSION: The performance of this method is significantly higher than the simple strategy consisting in transferring the annotation from the highest scoring BLAST match and is expected to find applications in automated functional annotation pipelines.
💡 Research Summary
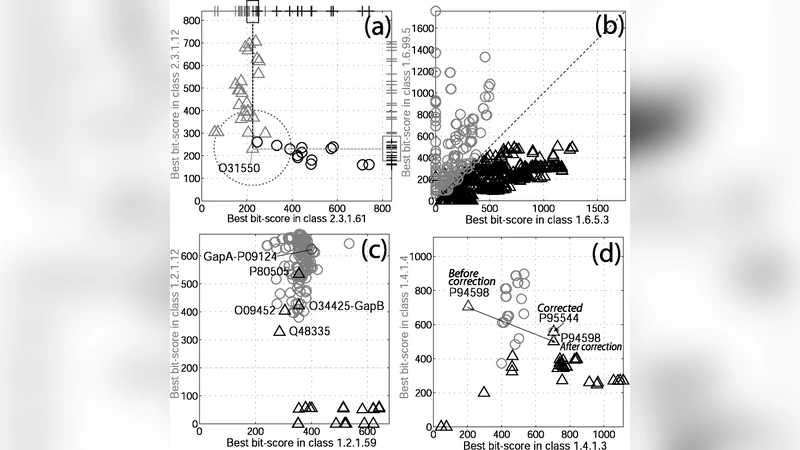
The paper introduces a novel Bayesian framework for protein function annotation that reverses the conventional workflow of clustering sequences first and then assigning functions. Instead of building clusters of homologous proteins and mapping functions onto those clusters, the authors start with a database of proteins already labeled according to a functional classification scheme—in this case, enzymes annotated with four‑digit EC numbers. Using sequence similarity searches, they compute “Correspondence Indicators” (CIs) that quantify the relationship between an uncharacterized query sequence and each functional class. These indicators can incorporate various search parameters such as alignment scores, alignment length, and E‑values, allowing flexible parametrisation of different similarity search strategies.
Two Bayesian models are formulated. The first model treats a single CI as the evidence and calculates posterior probabilities for class membership using a simple Bayes rule with class priors derived from the labeled database. The second model extends this to a multivariate setting, combining multiple CIs within a Bayesian network to capture more nuanced evidence and improve discrimination. Crucially, the Bayesian formulation yields explicit error‑rate estimates for each predicted class, providing a direct measure of annotation confidence that most existing pipelines lack.
The authors validate the approach on a curated set of over 50,000 enzyme sequences. For each query, they perform a BLAST search against the labeled database, compute CIs, and feed them into the Bayesian models. Performance is evaluated against the baseline strategy of transferring the annotation from the highest‑scoring BLAST hit. Across three key metrics—accuracy, recall, and estimated error rate—the Bayesian methods outperform the BLAST‑only approach. Notably, the advantage is most pronounced for sequences with moderate similarity, where simple top‑hit transfer often misclassifies. Detailed case studies illustrate instances where the Bayesian model assigns low posterior probabilities to dubious matches, correctly flagging potential errors.
The discussion emphasizes that mapping sequences directly to functional classes, rather than mapping functions onto clusters, simplifies the annotation pipeline and yields probabilistic confidence scores that can be integrated into automated workflows. The Bayesian framework is inherently extensible: new functional ontologies, additional sequence features, or alternative similarity metrics can be incorporated without redesigning the core algorithm. The authors suggest broad applicability to metagenomic projects, large‑scale protein databases, and real‑time annotation services. Future work is proposed to test the method on other classification schemes (e.g., Gene Ontology), handle multi‑label scenarios, and integrate the approach into existing annotation platforms to provide users with transparent, statistically grounded function predictions.
Comments & Academic Discussion
Loading comments...
Leave a Comment