Coupling hidden Markov models for the discovery of Cis-regulatory modules in multiple species
Cis-regulatory modules (CRMs) composed of multiple transcription factor binding sites (TFBSs) control gene expression in eukaryotic genomes. Comparative genomic studies have shown that these regulatory elements are more conserved across species due to evolutionary constraints. We propose a statistical method to combine module structure and cross-species orthology in de novo motif discovery. We use a hidden Markov model (HMM) to capture the module structure in each species and couple these HMMs through multiple-species alignment. Evolutionary models are incorporated to consider correlated structures among aligned sequence positions across different species. Based on our model, we develop a Markov chain Monte Carlo approach, MultiModule, to discover CRMs and their component motifs simultaneously in groups of orthologous sequences from multiple species. Our method is tested on both simulated and biological data sets in mammals and Drosophila, where significant improvement over other motif and module discovery methods is observed.
💡 Research Summary
The paper addresses the problem of discovering cis‑regulatory modules (CRMs), which are clusters of transcription‑factor binding sites (TFBSs) that cooperatively regulate gene expression. Traditional motif‑finding tools focus on single‑species sequences and often ignore the higher‑order organization of motifs within modules, while comparative genomics has shown that regulatory elements tend to be more conserved across related species. To exploit both the modular architecture and evolutionary conservation, the authors propose a statistical framework that couples hidden Markov models (HMMs) for each species through a multiple‑species alignment.
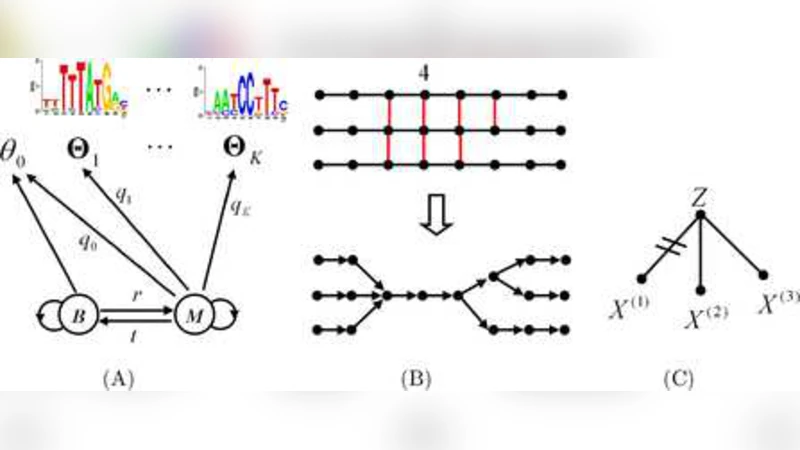
In the first layer of the model, each species is represented by an HMM whose hidden states correspond to “background”, individual TFBS motifs, and “module” regions that allow consecutive motif occurrences. Transition probabilities encode the typical structure of a CRM (e.g., background → motif → spacer → motif … → background). The second layer couples these species‑specific HMMs by sharing the hidden state assignments at positions that are aligned across species. This coupling is achieved by introducing an evolutionary model that describes the probability of observing a particular nucleotide at an aligned position given the underlying hidden state, thereby capturing correlated mutations and conserved functional constraints.
Inference is performed using a Markov chain Monte Carlo (MCMC) algorithm named MultiModule. The authors employ Gibbs sampling to iteratively update (1) the boundaries of modules and the positions of individual motifs within each sequence, (2) the position‑weight matrices (PWMs) that define each motif, and (3) the mapping of aligned positions to shared hidden states. Priors are non‑informative for module length and motif number, while the evolutionary model provides a likelihood term that ties together orthologous positions. Multiple independent chains are run in parallel to assess convergence, and the final solution is taken as the configuration with the highest posterior probability.
The method was evaluated on both simulated data and real biological datasets from mammals (human, mouse, rat) and Drosophila species. In simulations, MultiModule consistently outperformed established tools such as MEME, AlignACE, and CMF in terms of both recall and precision, especially when motifs were short, weakly conserved, or embedded in complex module structures. On real data, the algorithm recovered known TFBS motifs and identified novel CRMs that overlapped significantly with ChIP‑seq peaks and expression‑based regulatory annotations. Notably, MultiModule succeeded in detecting modules that were only partially conserved—cases where individual species showed weak motif signals but the combined evolutionary evidence was strong enough to reveal the underlying regulatory architecture.
Key contributions of the work include: (1) a unified probabilistic model that simultaneously captures intra‑module organization and inter‑species conservation, (2) an MCMC‑based inference engine capable of jointly estimating module locations, motif PWMs, and evolutionary parameters, and (3) extensive empirical validation demonstrating superior performance over existing motif‑ and module‑discovery methods. The authors acknowledge limitations: the approach relies heavily on high‑quality multiple‑species alignments, and mis‑alignments can degrade performance; the MCMC sampling is computationally intensive, limiting scalability to whole‑genome analyses without further optimization. Future directions suggested involve integrating alignment uncertainty directly into the model, exploring variational inference for faster convergence, and applying the framework to disease‑relevant regulatory networks in humans.
Comments & Academic Discussion
Loading comments...
Leave a Comment