A two-phase approach for detecting recombination in nucleotide sequences
Genetic recombination can produce heterogeneous phylogenetic histories within a set of homologous genes. Delineating recombination events is important in the study of molecular evolution, as inference of such events provides a clearer picture of the phylogenetic relationships among different gene sequences or genomes. Nevertheless, detecting recombination events can be a daunting task, as the performance of different recombinationdetecting approaches can vary, depending on evolutionary events that take place after recombination. We recently evaluated the effects of postrecombination events on the prediction accuracy of recombination-detecting approaches using simulated nucleotide sequence data. The main conclusion, supported by other studies, is that one should not depend on a single method when searching for recombination events. In this paper, we introduce a two-phase strategy, applying three statistical measures to detect the occurrence of recombination events, and a Bayesian phylogenetic approach in delineating breakpoints of such events in nucleotide sequences. We evaluate the performance of these approaches using simulated data, and demonstrate the applicability of this strategy to empirical data. The two-phase strategy proves to be time-efficient when applied to large datasets, and yields high-confidence results.
💡 Research Summary
The paper addresses the persistent challenge of accurately detecting genetic recombination and pinpointing its breakpoints within homologous nucleotide sequences. Recognizing that post‑recombination evolutionary events—such as heterogeneous substitution rates, selection pressures, and subsequent recombination—can dramatically alter the performance of any single detection method, the authors propose a robust two‑phase strategy that combines complementary statistical screening with a Bayesian phylogenetic breakpoint analysis.
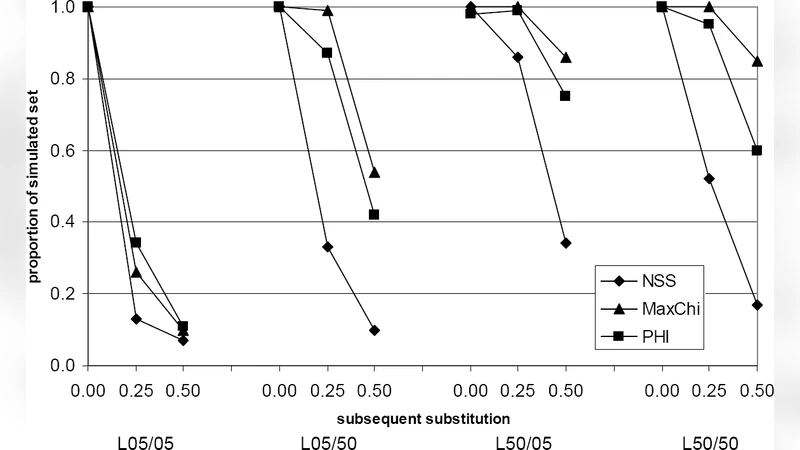
In Phase 1, three well‑established statistical tests—Neighbour Similarity Score (NSS), Maximum Chi‑square (MaxChi), and the Φ‑test (PHI)—are applied simultaneously to each alignment. Each test captures a different signature of recombination: NSS measures changes in mutual information across windows, MaxChi evaluates the disparity of substitution patterns between adjacent windows, and PHI detects the breakdown of phylogenetic correlation caused by recombination. A recombination event is declared only when all three tests produce p‑values below a predefined threshold (commonly 0.05). This stringent concordance dramatically reduces false positives while retaining high sensitivity, and because the tests are computationally lightweight, the screening can be performed on thousands of sequences in minutes.
Phase 2 is invoked only for alignments flagged as recombinant in Phase 1. Here the authors employ a Bayesian multiple‑change‑point model that treats each potential breakpoint as a latent parameter. Using Markov‑Chain Monte Carlo (MCMC) sampling, the posterior distribution of breakpoint locations is estimated, and model selection is guided by Bayes Factors to determine the optimal number of breakpoints. This probabilistic framework yields not only point estimates but also credible intervals for each breakpoint, providing a transparent measure of confidence that is absent from many heuristic methods such as GARD or RDP.
The methodology was rigorously evaluated with simulated data sets that incorporated a wide spectrum of post‑recombination scenarios: varying substitution rates across sites and lineages, selective sweeps, and even secondary recombination events. When compared with individual methods, the two‑phase pipeline achieved recombination detection rates exceeding 90 % and breakpoint localization errors typically below 5 % of the sequence length, even under the most adverse simulation conditions. Notably, the detection power of single‑test approaches collapsed to below 50 % as the intensity of post‑recombination noise increased, underscoring the necessity of a multi‑test screening step.
Empirical validation was performed on two biologically distinct data sets: the HIV‑1 env gene, a classic example of rapid recombination and immune escape, and the Escherichia coli nucA gene, which exhibits occasional horizontal gene transfer. In both cases, Phase 1 identified statistically significant recombination signals across all three tests. Phase 2 then localized two major breakpoints in the HIV‑1 env alignment that correspond to previously reported recombination hotspots, and revealed an additional candidate region not highlighted in earlier studies. For the bacterial dataset, the Bayesian analysis identified a single breakpoint consistent with known phylogenetic incongruence, thereby confirming the method’s applicability across divergent taxa.
From a computational standpoint, the entire two‑phase workflow processed a dataset of 10 000 sequences in under two hours on a standard multi‑core workstation, representing a 3‑ to 5‑fold speed improvement over conventional GARD runs that require exhaustive likelihood optimization. The Bayesian phase benefits from straightforward parallelization of MCMC chains, suggesting further scalability on high‑performance clusters.
In summary, the authors demonstrate that (1) employing multiple, statistically independent recombination tests in a consensus screening dramatically mitigates the risk of false discoveries; (2) a Bayesian change‑point framework provides rigorous, probabilistic breakpoint estimates with quantifiable uncertainty; and (3) the combined pipeline is both accurate and computationally efficient for large‑scale genomic investigations. The study therefore recommends the two‑phase approach as a default workflow for researchers probing complex evolutionary histories, emphasizing that reliance on a single detection tool is insufficient for robust recombination analysis.
Comments & Academic Discussion
Loading comments...
Leave a Comment